Software Citation:
Paper Citation: Jennifer Marie Shelton, Michelle C Coleman, Nic Herndon, Nanyan Lu, Ernest T Lam, Thomas Anantharaman, Palak Sheth, Susan J Brown. Tools and pipelines for BioNano data: molecule assembly pipeline and FASTA super scaffolding tool. bioRxiv doi: http://dx.doi.org/10.1101/020966
scripts to parse IrysView output
#Pipelines for BioNano data

The K-INBRE Bioinformatics Core has created easy to use pipelines for BioNano molecule maps or pre-assembled BioNano genome maps for several common assembly and/or alignment experiments.
All pipelines have sample datasets and tutorials. Pipelines take you from either raw data received from your mapping facility or assembled genome maps to finished analysis.
No experience with command line is necessary before using these scripts.
##Pipeline 1: Sewing Machine pipeline: iteratively super scaffold genome FASTA files with BioNano genome maps using stitch.pl
The sewing machine pipeline iteratively super scaffolds genome FASTA files with BioNano genome maps using stitch.pl and the BioNano tool RefAligner until no new super scaffolds can be produced. The pipeline runs alignments with both default and relaxed parameters. These alignments are then used by stitch.pl to superscaffold a fragmented genome FASTA. See tutorial lab to run the sewing machine pipeline with sample data https://github.com/i5K-KINBRE-script-share/Irys-scaffolding/blob/master/KSU_bioinfo_lab/stitch/sewing_machine_LAB.md.
##Pipeline 2: "Raw data-to-finished assembly and assembly analysis" pipeline for BioNano molecule maps with a sequence-based genome FASTA
The assemble XeonPhi pipeline preps raw molecule maps and writes and runs a series of assemblies for them. Then the user selects the best assembly and uses this to super scaffold the reference FASTA genome file and summarize the final assembly metrics and alignments.
The basic steps are to first merge multiple BNXs from a single directory and plot single molecule map quality metrics. Then rescale single molecule maps and plot rescaling factor per scan if reference is available. The rescaling step is analogous to the former "adjusting stretch scan by scan step". Next it writes scripts for assemblies with a range of parameters. After assemblies finish assembly metrics are generated and the best results are analyzed.
NOTE: This pipeline uses the same basic workflow as AssembleIrys.pl and AssembleIrysCluster.pl but it runs a Xeon Phi server with 576 cores (48x12-core Intel Xeon CPUs), 256GB of RAM, and Linux CentOS 7 operating system. Customization may be required to run the BioNano Assembler on a different machine. Specifically, customization of the "Customize RefAligner Settings" section of Irys-scaffolding/KSU_bioinfo_lab/assemble_XeonPhi/rescale_stretch.pl may be required to run the BioNano Assembler on a different machine. Customization of Irys-scaffolding/KSU_bioinfo_lab/assemble_XeonPhi/clusterArguments.xml may also be required for assembly to run successfully on a different cluster.
See tutorial lab to run the assemble XeonPhi pipeline with sample data https://github.com/i5K-KINBRE-script-share/Irys-scaffolding/blob/master/KSU_bioinfo_lab/assemble_XeonPhi/assemble_XeonPhi_LAB.md.
##Pipeline 3: "Raw data-to-finished de novo assembly and assembly analysis" pipeline for BioNano molecule maps
The assemble XeonPhi de novo pipeline preps raw molecule maps and writes and runs a series of assemblies for them. Then the user selects the best assembly then summarizes the final assembly metrics.
The basic steps are to first merge multiple BNXs from a single directory and plot single molecule map quality metrics. Next it writes scripts for assemblies with a range of parameters. After assemblies finish assembly metrics are generated and the best results are analyzed.
NOTE: This pipeline uses the same basic workflow as AssembleIrys.pl and AssembleIrysCluster.pl but it runs a Xeon Phi server with 576 cores (48x12-core Intel Xeon CPUs), 256GB of RAM, and Linux CentOS 7 operating system. Customization may be required to run the BioNano Assembler on a different machine. Specifically, customization of the "Customize RefAligner Settings" section of Irys-scaffolding/KSU_bioinfo_lab/assemble_XeonPhi/rescale_stretch.pl may be required to run the BioNano Assembler on a different machine. Customization of Irys-scaffolding/KSU_bioinfo_lab/assemble_XeonPhi/clusterArguments.xml may also be required for assembly to run successfully on a different cluster.
See tutorial lab to run the assemble XeonPhi pipeline with sample data https://github.com/i5K-KINBRE-script-share/Irys-scaffolding/blob/master/KSU_bioinfo_lab/assemble_XeonPhi/assemble_XeonPhi_de_novo_LAB.md.
#Other material and scripts for BioNano data
###intro_material
Examples of how work with BioNano software from the KSU Bioinformatics Core.
FAQs - Discussions or code that we are frequently asked about. Many of our tools are built out of smaller scripts that can also be used by themselves in the FAQ markdown we discuss things like generating an XMAP filtered by minimum percent alignment and what a CMAP is.
Windows_in_silico_labeling.md - How to install software on a Windows machine and videos of how to in silico label sequence data for alignment or to determine which enzymes to use for a BioNano project.
code_examples.sh - these are usage notes and general steps taken by the KSU Bioinformatics Core to assemble molecules or align assemblies. These can be used as a template for your own experiments with your BioNano data in a Linux environment.
IrysView_Troubleshooting.pdf - instructions on how to view all labels in an alignment if they do not automatically load.
cmap_stats.pl - Script outputs count of cmaps, cumulative lengths of cmaps and N50 of cmaps. Tested on CMAP File Version: 0.1.
perl ~/Irys-scaffolding/KSU_bioinfo_lab/map_tools/cmap_stats.pl -c ~/Irys-scaffolding/KSU_bioinfo_lab/sample_output_directory/BioNano_consensus_cmap/ESCH_COLI_1_2015_000_STRICT_T_150_REFINEFINAL1.cmap
xmap_stats.pl - Script outputs breadth of alignment coverage and total aligned length from an xmap. Tested on XMAP File Version: 0.1. "Breadth of alignment coverage" is the number of bases covered by aligned maps. This is equivalent to "Total Unique Aligned Len(Mb)". "Total alignment length is the total length of the alignment. This is equivalent to "Total Aligned Len(Mb)".
perl ~/Irys-scaffolding/KSU_bioinfo_lab/map_tools/xmap_stats.pl -x ~/Irys-scaffolding/KSU_bioinfo_lab/sample_output_directory/align_in_silico_xmap/NC_010473_mock_scaffolds_to_ESCH_COLI_1_2015_000_STRICT_T_150_REFINEFINAL1.xmap
bnx_stats.pl - Script outputs count of molecule maps in BNX files, cumulative lengths of molecule maps and N50 of molecule maps. Script also outputs a PDF with these metrics as well as histograms of molecule map quality metrics. Tested on BNX File Version 1.0 and Version 1.2. The user inputs a list of BNX files or a glob as the final arguments to script. Users can filter results by min molecule length in kb using the -l flag. Things to add include switching between QC and cleaning.
Script has no options other than help menus and min length currently but it was designed to be adapted into a molecule cleaning script similar to prinseq or fastx. Feel free to fork this and add your own filters.
perl ~/Irys-scaffolding/KSU_bioinfo_lab/map_tools/bnx_stats.pl -l 150 /home/bionano/Irys-scaffolding/KSU_bioinfo_lab/sample_output_directory/Datasets/*/*/Molecules.bnx
hybridScaffold_finish_fasta.pl - Script creates new FASTA files including new hybrid sequences output by hybridScaffold and all sequences that were not used by hybridScaffold with their original headers. Also outputs a text file list of the headers for sequences that were used to make the new hybrid sequences.
perl hybridScaffold_finish_fasta.pl -x HYBRID_SCAFFOLD.xmap -s HYBRID_SCAFFOLD.fasta -f original.fasta
CmapById.pl - Script outputs new CMAP with only maps with user specified IDs. Tested on CMAP File Version: 0.1. Call with "-help" flag for detailed instructions.
perl ~/Irys-scaffolding/KSU_bioinfo_lab/map_tools/CmapById.pl -c sample_data/sample.cmap -i 1,3,6 -o sample_data/sample_out_file
perl ~/Irys-scaffolding/KSU_bioinfo_lab/map_tools/CmapById.pl -c sample_data/sample.cmap -i 3..10 -o sample_data/sample_out_3_10
perl ~/Irys-scaffolding/KSU_bioinfo_lab/map_tools/CmapById.pl -c sample_data/sample.cmap -i 2 -o sample_data/sample_out_cmap_2
flip.pl - This utility script reads from a list of maps to flip from a txt file (one CMAP id per line) and creates a CMAP with the requested flips.
stitch.pl - a package of scripts that analyze IrysView output (i.e. XMAPs). The script filters XMAPs by confidence and the percent of the maximum potential length of the alignment and generates summary stats of the more stringent alignments. The first settings for confidence and the minimum percent of the full potential length of the alignment should be set to include the range that the researcher decides represent high quality alignments after viewing raw XMAPs. Some alignments have lower than optimal confidence scores because of low label density or short sequence-based scaffold length. The second set of filters should have a user-defined lower minimum confidence score, but a much higher percent of the maximum potential length of the alignment in order to capture these alignments. Resultant XMAPs should be examined in IrysView to see that the alignments agree with what the user would manually select.
stitch.pl finds the best super-scaffolding alignments each run. It can be run iteratively until all super-scaffolds have been found by creating a new cmap from the output super-scaffold fasta, aligning this cmap as the query with the BNG consensus map as the reference and using the x_map, r_cmap and the super-scaffold fasta as input for another run of stitch.pl. See KSU_bioinfo_lab/stitch for more details.
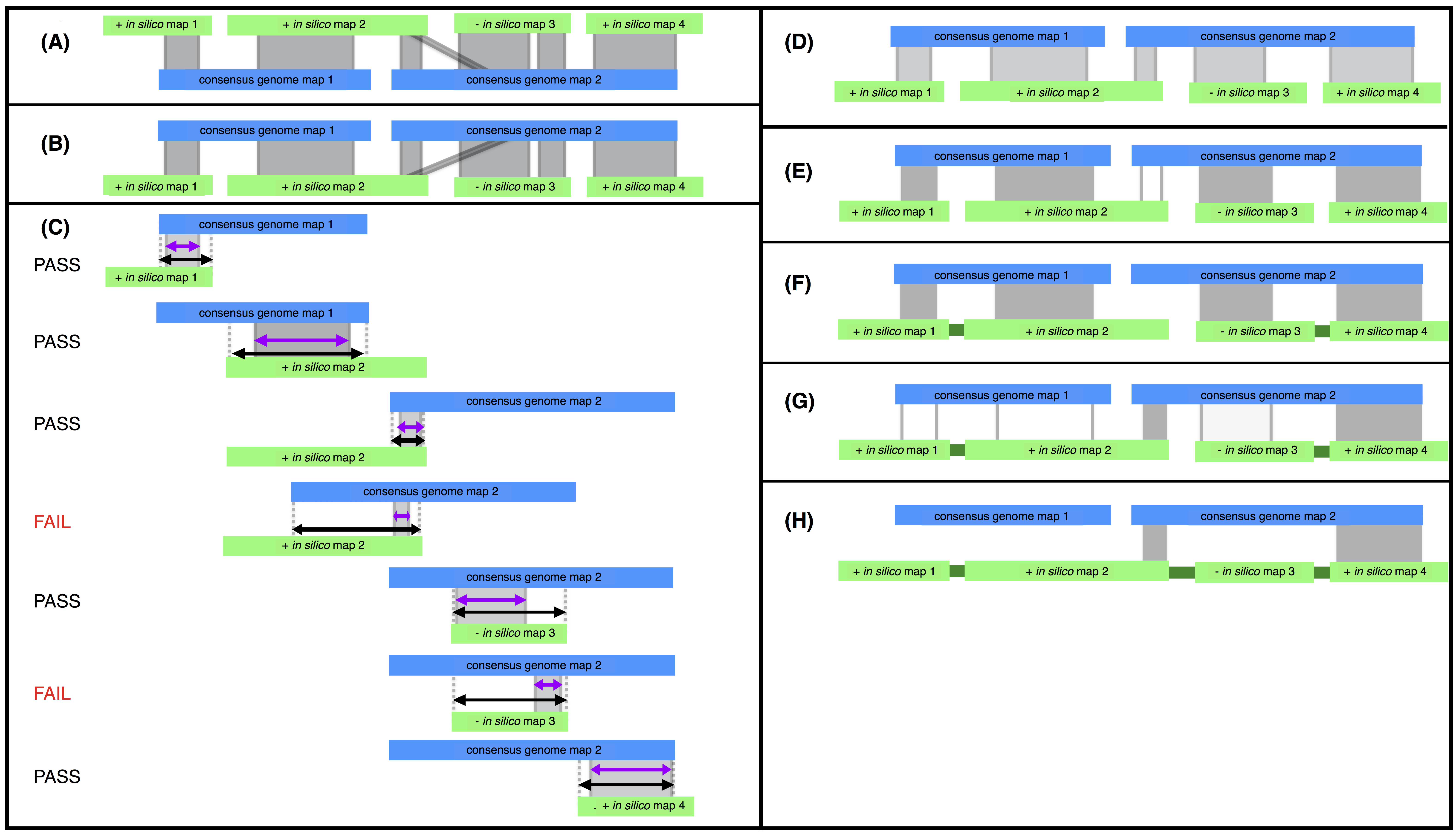
Steps of the stitch.pl algorithm. Consensus genome maps (blue) are shown aligned to in silico maps (green). Alignments are indicated with grey lines. CMAP orientation for in silico maps is indicated with a ”+” or ”-” for positive or negative orientation respectively. (A) The in silico maps are used as the reference. (B) The alignment is inverted and used as input for stitch.pl. (C) The alignments are filtered based on alignment length (purple) relative to total possible alignment length (black) and confidence. Here assuming all alignments have high confidence scores and the minimum percent aligned is 40% two alignments fail for aligning over less than 40% of the potential alignment length for that alignment. (D) Filtering produces an XMAP of high quality alignments with short (local) alignments removed. (E) High quality scaffolding alignments are filtered for longest and highest confidence alignment for each in silico map. The third alignment (unshaded) is filtered because the second alignment is the longest alignment for in silico map 2. (F) Passing alignments are used to super scaffold (captured gaps indicated in dark green). (G) Stitch is iterated and additional super scaffolding alignments are found using second best scaffolding alignments. (H) Iteration takes advantage of cases where in silico maps scaffold consensus genome maps as in silico map 2 does. Stitch is run iteratively until all super scaffolding alignments are found.
DEPENDENCIES
git - see http://git-scm.com/book/ch1-4.html for instructions
BioPerl - see http://www.bioperl.org/wiki/Installing_BioPerl
Requires BNGCompare from https://github.com/i5K-KINBRE-script-share/BNGCompare in your home
directory. Also requires RefAligner. Install BioNano scripts and
executables in `~/scripts` and `~/tools` directories respectively. Follow the Linux installation
instructions in the "2.5.1 IrysSolve server RefAligner and Assembler" section of
http://www.bnxinstall.com/training/docs/IrysViewSoftwareInstallationGuide.pdf to install
RefAligner.
USAGE
See README for more details https://github.com/i5K-KINBRE-script-share/Irys-scaffolding/blob/master/KSU_bioinfo_lab/stitch/README.md
Test with sample datasets
See tutorial lab to run the sewing machine pipeline with sample data https://github.com/i5K-KINBRE-script-share/Irys-scaffolding/blob/master/KSU_bioinfo_lab/stitch/sewing_machine_LAB.md.
NOTE: AssembleIrysCluster.pl is no longer supported. This workflow has been replaced by AssembleIrysXeonPhi.pl. AssembleIrysXeonPhi.pl runs on a Xeon Phi server with 576 cores (48x12-core Intel Xeon CPUs), 256GB of RAM, and Linux CentOS 7 operating system. See the following tutorials for details on the new workflow:
SUMMARY
AssembleIrysCluster.pl - Adjusts stretch by scan. Merges BNXs and writes scripts for assemblies with a range of parameters. This script uses the same workflow as AssembleIrys.pl but it runs on the Beocat SGE cluster.
Workflow diagram
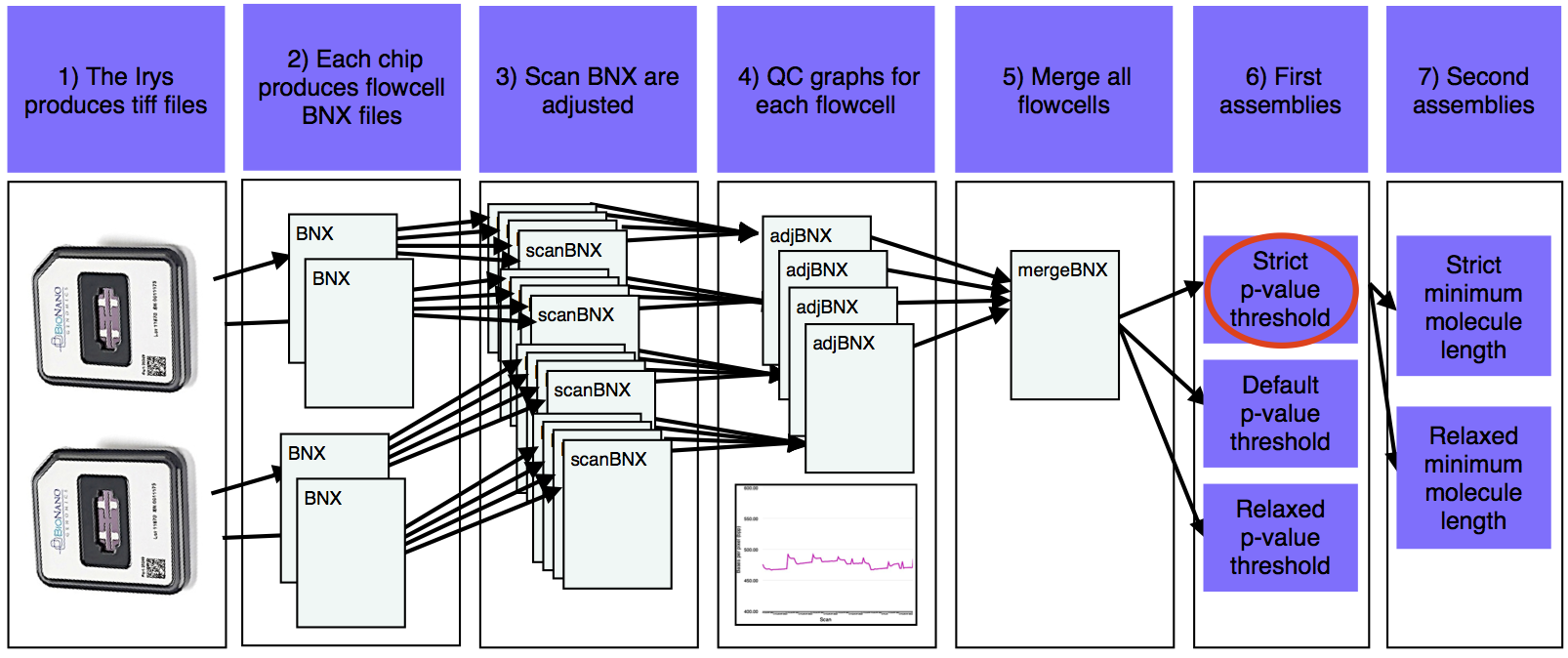
- The Irys produces tiff files that are converted into BNX text files.
- Each chip produces one BNX file for each of two flowcells.
- BNX files are split by scan and aligned to the sequence reference. Stretch (bases per pixel) is recalculated from the alignment.
- Quality check graphs are created for each pre-adjusted flowcell BNX.
- Adjusted flowcell BNXs are merged.
- The first assemblies are run with a variety of p-value thresholds.
- The best of the first assemblies (red oval) is chosen and a version of this assembly is produced with a variety of minimum molecule length filters.
USAGE
perl AssembleIrysCluster.pl -g [genome size in Mb] -r [reference CMAP] -b [directory with BNX files] -p [project name]
DEPENDENCIES
Perl module XML::Simple. This can be installed using CPAN http://search.cpan.org/~grantm/XML-Simple-2.20/lib/XML/Simple.pm;
Perl module Data::Dumper. This can be installed using CPAN http://search.cpan.org/~smueller/Data-Dumper-2.145/Dumper.pm;
assembly_qc.pl - a script that compiles assembly metrics for assemblies in all of the possible directories:'strict_t', 'default_t', 'relaxed_t', 'strict_t/strict_ml', 'strict_t/relaxed_ml', 'default_t/strict_ml', 'default_t/relaxed_ml', 'relaxed_t/strict_ml', and 'relaxed_t/relaxed_ml'. The assemblies are created using assemble_SGE_cluster/AssembleIrysCluster.pl from https://github.com/i5K-KINBRE-script-share/Irys-scaffolding/tree/master/KSU_bioinfo_lab. The parameter -b should be the same th same as the -b parameter used for the assembly script (same with the -p parameter). It is the directory with the BNX files used for assembly.
perl ~/Irys-scaffolding/KSU_bioinfo_lab/assembly_qc.pl -b ~/sample_data -p My_project_ID
###sv_detect
These scripts were created to generate BED files of gaps in sequence-based assemblies and to run sv_detect from BioNano as a standalone pipeline but the scripts may be redundant now with the new IrysView release. If we find that they are they will be removed.
Scripts in the Irys-scaffolding/KSU_bioinfo_lab/script_archive directory are former versions of workflows that are currently not supported.
###assemble/AssembleIrys.pl
SUMMARY
AssembleIrys.pl - Adjusts stretch by scan. Merges BNXs and initiate assemblies with a range of parameters. This script uses the same workflow as AssembleIrysCluster.pl but it runs on local Linux machines. This script has not been updated to account for frequent changes in Bionano output format. See AssembleIrysXeonPhi.pl for fequently updated scripts.
SUMMARY
analyze_irys_output.pl - This script was replaced by stitch.pl