Are you curious about what's the picture for?
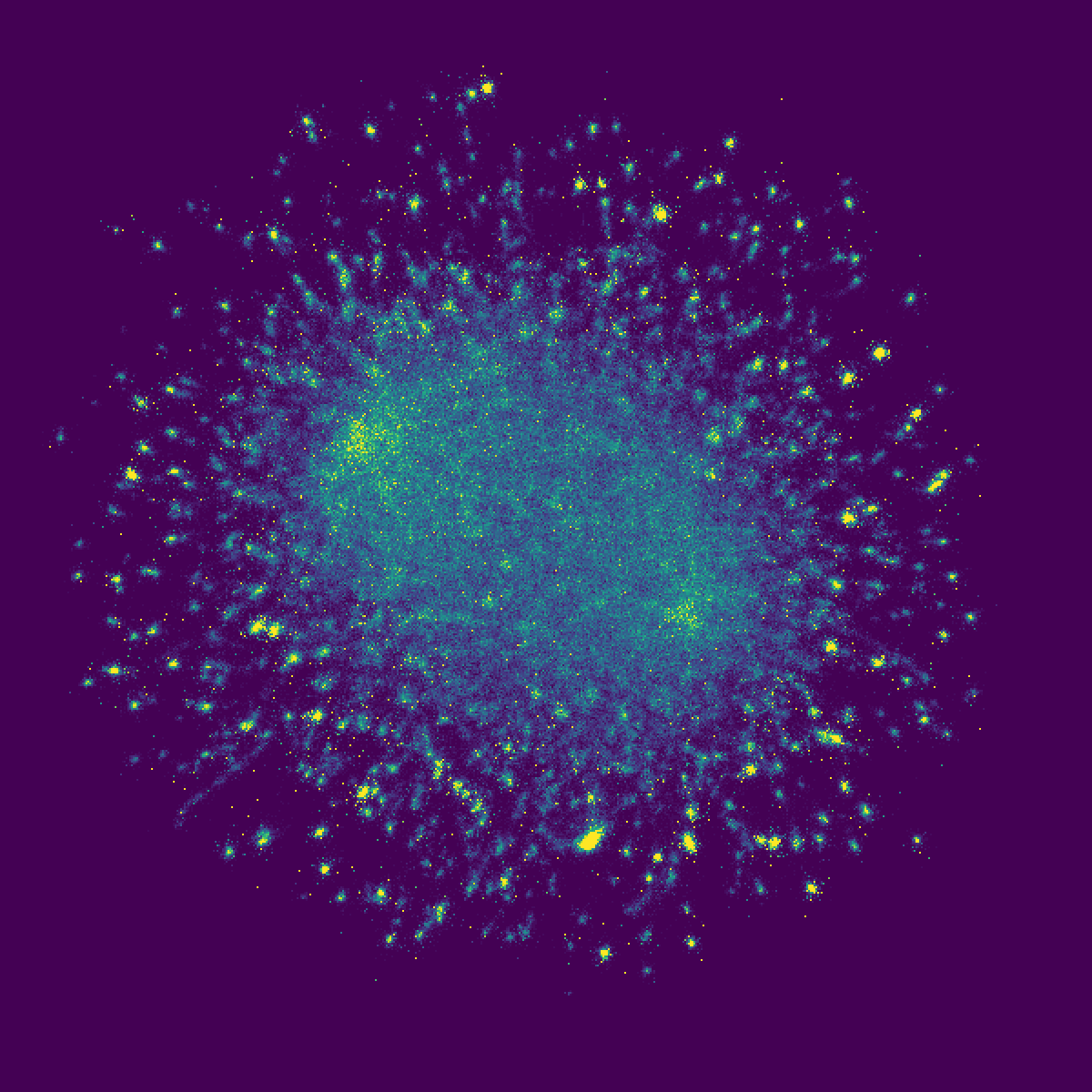
This amazing picture is from the UMAP projection of the 21-nt sequences of 999,240 human A-to-I RNA editing sites (non-Alu repeats) in REDIportal! Each pixel here represents a set of similar sequences, and brightness of the pixels represent the density of sites. With this picture, we can intuitively learn what kinds of k-mers (21-mers here) are included and enriched in such huge amount of RNA editing events. This is never been done before.
interactive epitranscriptomic Motif Visualization and Sub-type Partitioning (iMVP) is a strategy inspired by the commonly used single cell analysis strategy of dimensional reduction followed by clustering. Different from the digital counts in single cell analysis, we here use the RNA sequence (and/or structure) as an input. Here, we firstly transform RNA sequences into a one-hot format; then these transformed sequences were projected to a 2D plane with UMAP, which can not only gather the similar sequences together, but also maintain the relationships between each other (compared with t-SNE); the dimensional reduced data were further clustered by density, with the super-efficient algorithm HDBSCAN, to highlight the enriched sequences.
Compared with canonical motif discovery strategies, such as MEME (statistics driven), iMVP no longer relays on the given parameters of window lengths, p-value cutoff, and etc. It is more intuitive for motif finding and more sensitive for minor motifs, making the motif finding process transparent. More importantly, we are now able to analyze modification sites in a extremely huge number. For example, the largest dataset we analyzed is the human A-to-I editing records with >15,000,000 sites in >5,000,000 21-mers. We finished the analysis with a single RTX2080Ti GPU within 1 hour, which is impossible for any other motif finders.
A detailed document can be found here: https://imvp.readthedocs.io/en/latest/.

iMVP is currently not a single package. We provide two ways to apply iMVP in your analysis:
- For new users not experienced in Python, we suggest you to use our interactive interface for quick analysis.
- For advanced users, we suggest you to follow our
Notebooks(method-to-modifications):HeLa_Noc.ipynbandHuman_dev.ipynbare for the basics construction of an iMVP analysis (one-to-one).m6A_methods.ipynbandPseudoU_methods.ipynbare for the comparative analysis of different methods for RNA modification discovery (one-to-several).RNA_seq_var.ipynbis for modification discovery from RNA-seq variants (one-to-many).m6A_nanopore.ipynbis for phase-matching stratey for Nanopore modification discovery (one-to-many).Benchmarking_[xxx].ipynbare Notebooks for method and parameter selection guidances.
- More background knowledge for
UMAPandHDBSCANcan be found here (https://umap-learn.readthedocs.io/en/latest/) and here (https://hdbscan.readthedocs.io/en/latest/).
We use Python 3 in iMVP. We list the packages required in each notebook. In summary, we require pandas, numpy, umap-learn, hdbscan, scipy, matplotlib, and seaborn for most of the usages. For some cases, scikit-learn should be isntalled for benchmarking. We recommend you to install Weblogo and MEME for further analysis of the clusters.
For Linux (and probably for MacOS), all of the packages and softwares are available on Pypi (with pip) or on the official websites:
pip install iMVP-utils
We don't recommend running iMVP with Windows. Please note that, hdbscan from Pypi is not compatible, instead, you can get an wheels (https://www.lfd.uci.edu/~gohlke/pythonlibs/) with some troubles with multi-threads (please modify core_n_jobs if you get some troubles). In addition, Weblogo and MEME are not available on Windows, too.
To achieve analysis with huge dataset, we use NVIDIA RAPIDS library (majorly cuML) to accelerate UMAP and HDBSCAN. Please visit the office pages for more information.
Please note that, we can speed up UMAP with RRAPIDS in more than 40-folds. But it's hard for HDBSCAN, because we will have various computational complexity with different data and different parameters. I offen get out of GPU memory if too many small clusters are wating for analyzing. Hence, the current best practices for large dataset (e.g., 40 million non-repeative editing sites) is to use UMAP to compute the corrdinates on the 2D plane, then process the projections to CPU version HDBSCAN for further clustering. We also notice that, soft-clustering strategy will be buggy when the condensed tree is too large.
Please use iMVP_viewer.py to access the interactive interface.
- The idea, major codes, and notebooks are contributed by
Jianheng Liuat Rui Zhang's Lab (Jan, 2022). - The interactive version is contributed by
Jing YaoandJianheng Liuat Rui Zhang's lab (Feb, 2022).
MIT License
Copyright (c) 2022 JH Liu
Permission is hereby granted, free of charge, to any person obtaining a copy of this software and associated documentation files (the "Software"), to deal in the Software without restriction, including without limitation the rights to use, copy, modify, merge, publish, distribute, sublicense, and/or sell copies of the Software, and to permit persons to whom the Software is furnished to do so, subject to the following conditions:
The above copyright notice and this permission notice shall be included in all copies or substantial portions of the Software.
THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
For any bugs, please post them in Issues.
For the issues about iMVP framework, you can also contact Jiangheng Liu (jhfoxliu@gmail.com or jil4026@med.cornell.edu) directly.
For the issues about the interactive interface (bugs, feature requests, and etc.), please contact Jing Yao (yaoj39@mail2.sysu.edu.cn).
Liu, J., Huang, T., Yao, J. et al. Epitranscriptomic subtyping, visualization, and denoising by global motif visualization. Nat Commun 14, 5944 (2023). https://doi.org/10.1038/s41467-023-41653-4