This is wrapper for recuitment plot based on blast based on the recruitment plot function in enevoemics package. You mush have R and Perl installed on your machine. By default perl is installed on Linux and MacOS. You need to install R for plotting (R4.0.5 or higher is recommended). I want to thank Genevie for the first version. Please contact me jianshuzhao@yahoo.com
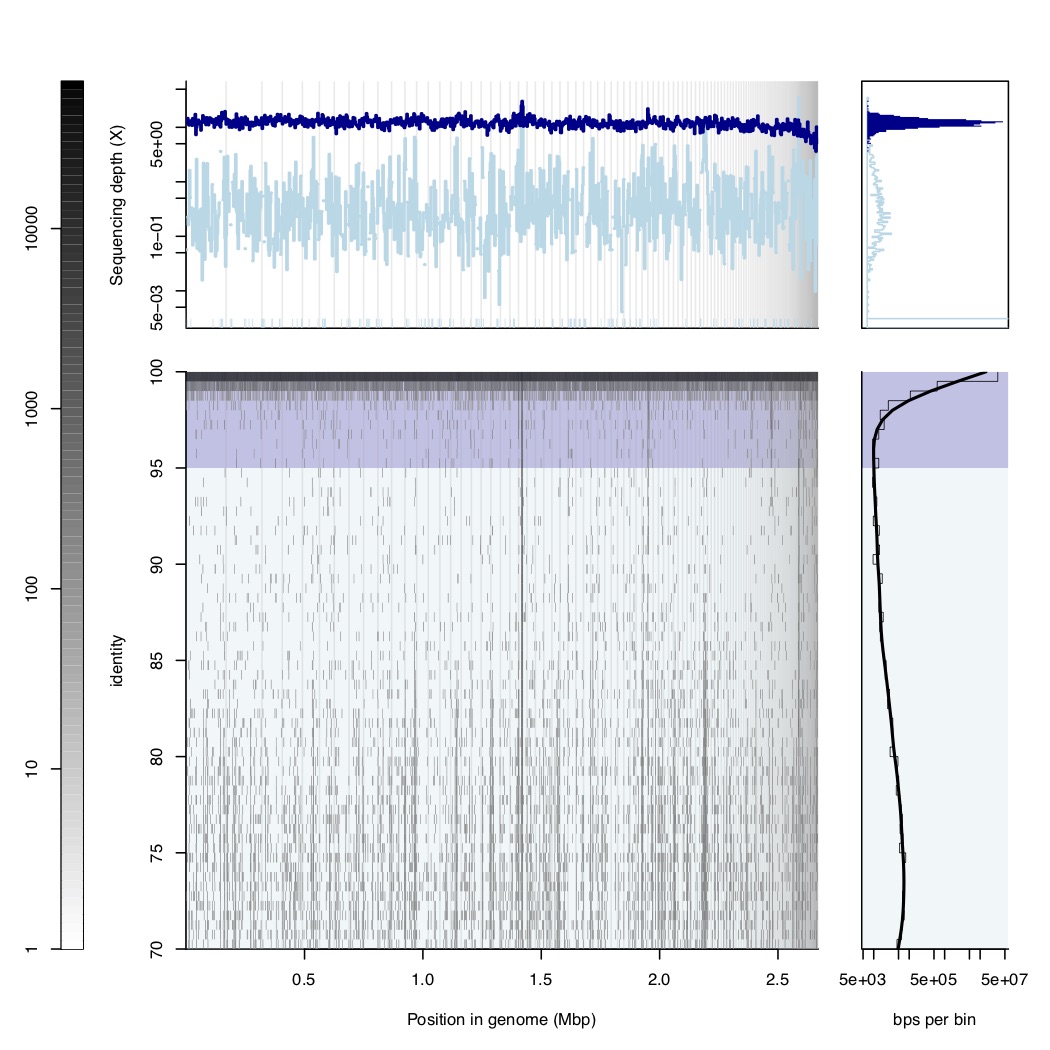
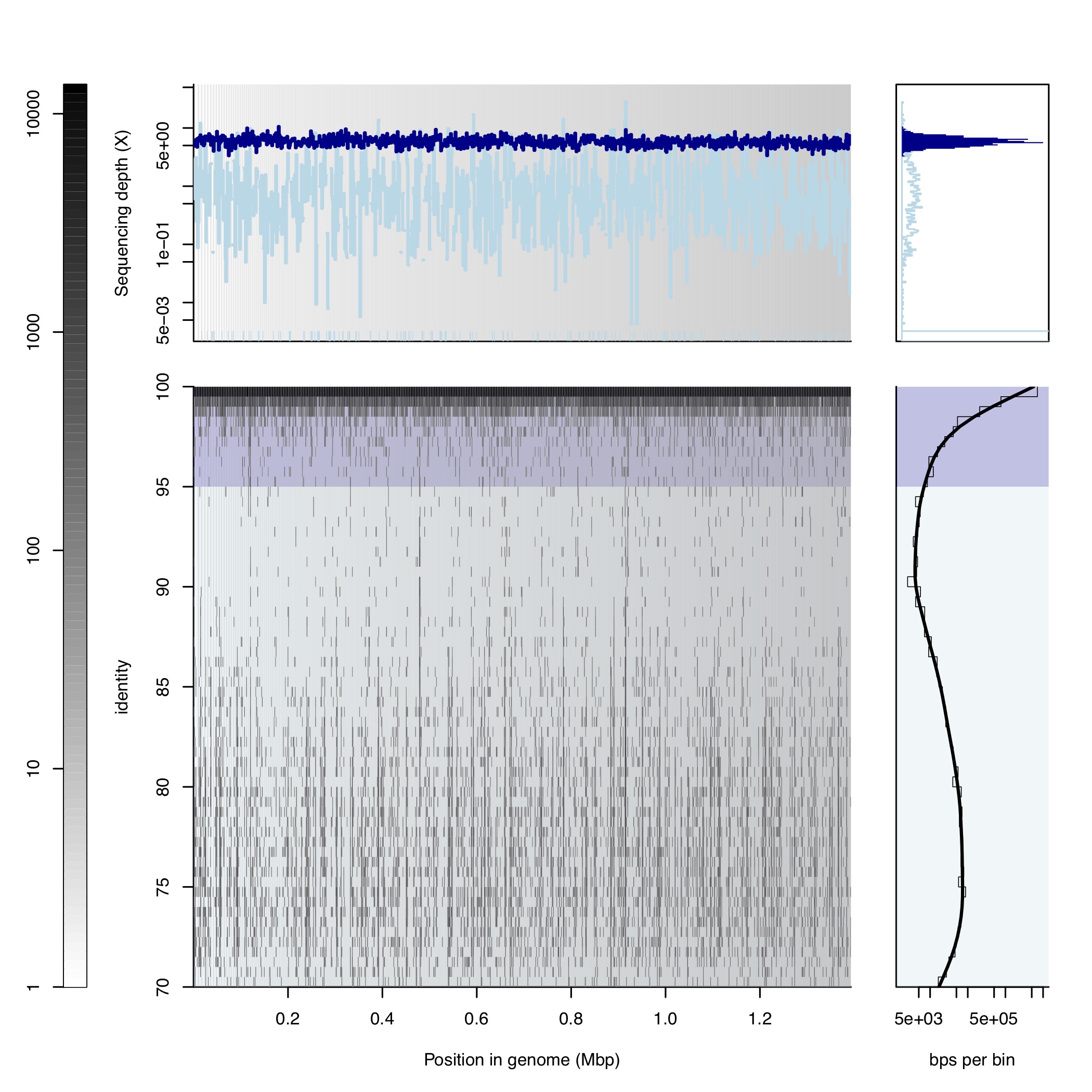
For any metagenome from environment, high quality genomes should have a reads mapping gap following blastN algorithm (-task blastn, slow but very sentitive at low identity regions), any other mappers like bwa, bowtie2 and minimaps will miss a lot of low identity reads especailly less than 80% identity so the reads mapping gap will disappear. The gap you see at the end of this page showed a great example of seuqences discret population (Aka, microbial population for a species). You must do competitive mapping for all MAGs you recovered from this sample with the blastN algorithm to see it.
You must have blast v2.12.0 for now because this version has a great imporvement of parallelism for large metagenomes. Early version will not be used and there will be an error.
You can create as many plots as you want parallelly. The blast step take some time. For a metagenomes with 3.0 GB (forwared reads only, unzipped) and 14 reference genomes. It take 2 hours.
blast+ and seqtk must be installed for alignment. If you are using conda, run conda install -c bioconda blast seqtk
Only Interleaved (people also call it merge in bbtools software) reads are supported for now. I will add support for forward and reverse reads.
If you have paired read, use the seqtk software to intleave your reads:
conda install seqtk -c bioconda
seqtk mergepe R1.fasta.gz R2.fasta.gz > interleaved.fasta
Usage: ./makeRecruitmentPlot.sh database_dir query.fa output_base
database_dir directory that contains fasta files (must ends with .fasta) which will be the database [most likely your longer sequence]
query.fa Fasta file that will be mapped to the database [most likely your reads]
output_base Base name for the blast output and recruitment plots
blast output: output_base.blst [Unique matches with over 70% coverage and 50 bp match]
recruitment object: output_base.recruitment.out
recruitment pdf: output_base.recruitment.pdf
## on Linux
git clone https://github.com/jianshu93/RecruitmentPlot_blast
cd RecruitmentPlot_blast
### Get example interleaved reads data mentioned above, genomes offered are binned and refine from this metagenome
wget http://rothlab.com/Data/T4AerOil_sbsmpl5.fa.gz
mv T4AerOil_sbsmpl5.fa.gz ./demo_input
gunzip ./demo_input/T4AerOil_sbsmpl5.fa.gz
### run the orginal blastn algorithm, which is very slow but very useful for check sequence discrete population
./makeRecruitmentPlot_linux_blastN.sh ./demo_input/MAG ./demo_input/T4AerOil_sbsmpl5.fa try
### run default fast mode (megablast)
./makeRecruitmentPlot_linux.sh ./demo_input/MAG ./demo_input/T4AerOil_sbsmpl5.fa try
## on MacOS, install homebrew first
brew install grep
brew install coreutils
git clone https://github.com/jianshu93/RecruitmentPlot_blast
cd RecruitmentPlot_blast
### Get example interleaved reads data mentioned above, genomes offered are binned and refine from this metagenome
wget http://rothlab.com/Data/T4AerOil_sbsmpl5.fa.gz
mv T4AerOil_sbsmpl5.fa.gz ./demo_input
gunzip ./demo_input/T4AerOil_sbsmpl5.fa.gz
./makeRecruitmentPlot.sh ./demo_input/MAG ./demo_input/T4AerOil_sbsmpl5.fa try
lab5_MAG_001 recruitment plot
lab5_MAG_002 recruitment plot
Rodriguez-R, Luis M. and Konstantinos T. Konstantinidis. 2016. “The Enveomics Collection: a Toolbox for Specialized Analyses of Microbial Genomes and Metagenomes.” PeerJ 1–16.