-
Notifications
You must be signed in to change notification settings - Fork 2
Commit
This commit does not belong to any branch on this repository, and may belong to a fork outside of the repository.
- Loading branch information
Katherine Eaton
committed
Aug 11, 2022
1 parent
b968dc6
commit 3d53ebd
Showing
2 changed files
with
101 additions
and
48 deletions.
There are no files selected for viewing
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -1,49 +1 @@ | ||
| # Development | ||
|
|
||
| ## Workflow | ||
|
|
||
| - Change parent clade nomenclature from `Omicron/21K` to `Omicron/BA.1/21K`. | ||
| - Identify false positives if the recombinant region doesn't have a unique substitution. | ||
| - Remove all subtree related rules (#52) | ||
| - Use latest protobufs rather than time-stamped (#54) | ||
| - Remove `sc2rf` debug mode. | ||
|
|
||
| ## Programs | ||
|
|
||
| - Upgrade `nextclade` from `v1.11.0` to `v2.2.0`. | ||
|
|
||
| ## Output | ||
|
|
||
| - Renamed `parents` column to `parents_clade`. | ||
| - Added `parents_lineage` column. | ||
| - Restrict `nextclade` output to `fasta,tsv` (alignment and QC table). | ||
| - Remove subtrees. | ||
|
|
||
| ## Params | ||
|
|
||
| - Tested out `--enable-deletions` for `sc2rf` again, caused issues for `XD`, as the first breakpoint no longer contains a breakpoint motif. | ||
| - Make `exclude_clades` param of `nextclade_recombinants` completely optional. | ||
| - Add `B.1.438.1` to `clades` for `proposed808`. | ||
| - Add `B.1.631` and `B.1.634` `clades` for XB. | ||
|
|
||
| ## Data | ||
|
|
||
| - Add proposed771 (BA.5,BA.2) to positive controls. | ||
| - Designated recombinants currently not included in controls: | ||
|
|
||
| - XA | OU143858.1, OU106081.1 | ||
| - XB | OK219662.1, OK292107.1 | ||
| - XC | No public genomes, restricted to Japan | ||
| - XK | No public genomes | ||
| - XT | No public genomes, restricted to South Africa | ||
| - XU | EPI_ISL_10523603 | ||
| - XV | OX099731.1, OX097667.1 | Restricted to Denmark and Italy | ||
| - XY | OW680552.1, ON350451.1 | ||
| - XZ | ON388919.1, ON591206.1 | ||
| - XAB | EPI_ISL_11300019 | ||
| - XAC | EPI_ISL_12683057 | ||
| - XAD | EPI_ISL_12523233 | ||
| - XAE | ON034711.1, OM999631.1 | ||
| - XAF | ON588970.1, ON674975.1 | ||
| - XAG | EPI_ISL_13068320 | ||
| - XAH | EPI_ISL_13253581 |
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,101 @@ | ||
| # # v0.4.0 | ||
|
|
||
| ## General | ||
|
|
||
| v0.4.0 has been trained and validated on the latest generation of SARS-CoV-2 Omicron clades (ex. 22A/BA.4 and 22B/BA.5). Recombinant sequences involving BA.4 and BA.5 can now be detected, unlike in v0.3.0 where they were not included in the `sc2rf` models. | ||
|
|
||
| v0.4.0 is also a **major** update to how sequences are categorized into lineages/clusters. A recombinant lineage is now defined as a group of sequences with a unique combination of: | ||
|
|
||
| - Lineage assignment (ex. `XM`) | ||
| - Parental clades (ex. `Omicron/21K,Omicron/21L`) | ||
| - Breakpoints (ex. `17411:21617`) | ||
| - **NEW**: Parental lineages (ex. `BA.1.1,BA.2.12.1`) | ||
|
|
||
| Novel recombinants (i.e. undesignated) can be identified by a lineage assignment that does not start with `X*` (ex. `BA.1.1`) _or_ with a lineage assignment that contains `-like` (ex. `XM-like`). A cluster of sequences may be flagged as `-like` if one of the following criteria apply: | ||
|
|
||
| 1. The lineage assignment by [Nextclade](https://github.com/nextstrain/nextclade) conflicts with the published breakpoints for a designated lineage (`resources/breakpoints.tsv`). | ||
|
|
||
| - Ex. An `XE` assigned sample has breakpoint `11538:12879`, which conflicts with the published `XE` breakpoint (`ex. 8394:12879`). This will be renamed `XE-like`. | ||
|
|
||
| 1. The cluster has 10 or more sequences, which share at least 3 private mutations in common. | ||
|
|
||
| - Ex. A large cluster of sequences (N=50) are assigned `XM`. However, these 50 samples share 5 private mutations `T2470C,C4586T,C9857T,C12085T,C26577G` which do not appear in true `XM` sequences. These will be renamed `XM-like`. Upon further review of the reported matching [pango-designation issues](https://github.com/cov-lineages/pango-designation/issues) (`460,757,781,472,798`), we find this cluster to be a match to `proposed798`. | ||
|
|
||
| The ability to identify parental lineages and private mutations is largely due to improvements in the [newly released nextclade datasets](https://github.com/nextstrain/nextclade_data/releases/tag/2022-07-26--13-04-52--UTC), , which have increased recombinant lineage accuracy. As novel recombinants can now be identified without the use of the custom UShER annotations (ex. proposed771), all UShER rules and output have been removed. This significantly improves runtime, and reduces the need to drop non-recombinant samples for performance. The result is more comparable output between different dataset sizes (4 samples vs. 400,000 samples). | ||
|
|
||
| > **Note!** Default parameters have been updated! Please regenerate your profiles/builds with: | ||
| > | ||
| > ```bash | ||
| > scripts/create_profile.sh --data data/custom | ||
| > ``` | ||
| ## Datasets | ||
|
|
||
| - [Issue #49](https://github.com/ktmeaton/ncov-recombinant/issues/49): The tutorial lineages were changed from `XM`,`proposed467`, `miscBA1BA2Post17k`, to `XD`, `XH`, `XAN`. The previous tutorial sequences had genome quality issues. | ||
| - [Issue #51](https://github.com/ktmeaton/ncov-recombinant/issues/51): Add `XAN` to the controls dataset. This is BA.2/BA.5 recombinant. | ||
| - [Issue #62](https://github.com/ktmeaton/ncov-recombinant/issues/62): Add `XAK` to the controls dataset. This is BA.2/BA.1 VUM recombinant monitored by the ECDC. | ||
|
|
||
| ## Nextclade | ||
|
|
||
| - [Issue #46](https://github.com/ktmeaton/ncov-recombinant/issues/46): `nextclade` is now run twice. Once with the regular `sars-cov-2` dataset and once with the `sars-cov-2-no-recomb` dataset. The `sars-cov-2-no-recomb` dataset is used to get the nucleotide substitutions before recombination occurred. These are identified by taking the `substitutions` column, and excluding the substitutions found in `privateNucMutations.unlabeledSubstitutions`. The pre-recombination substitutions allow us to identify the parental lineages by querying [cov-spectrum](https://cov-spectrum.org/). | ||
| - [Issue #48](https://github.com/ktmeaton/ncov-recombinant/issues/48): Make the `exclude_clades` completely optional. Otherwise an error would be raised if the user didn't specify any. | ||
| - [Issue #50](https://github.com/ktmeaton/ncov-recombinant/issues/50): Upgrade from `v1.11.0` to `v2.3.0`. Also upgrade the default dataset tags to [2022-07-26T12:00:00Z](https://github.com/nextstrain/nextclade_data/releases/tag/2022-07-26--13-04-52--UTC) which had significant bug fixes. | ||
| - [Issue #51](https://github.com/ktmeaton/ncov-recombinant/issues/51): Relax the recombinant criteria, by flagging sequences with ANY labelled private mutations as a potential recombinant for further downstream analysis. This was specifically for BA.5 recombinants (ex. `XAN`) as no other columns from the `nextclade` output indicated this could be a recombinant. | ||
| - Restrict `nextclade` output to `fasta,tsv` (alignment and QC table). This saves on file storage, as the other default output is not used. | ||
|
|
||
| ## sc2rf | ||
|
|
||
| - [Issue #51](https://github.com/ktmeaton/ncov-recombinant/issues/51): `sc2rf` is now run twice. First, to detect recombination between clades (ex. `Delta/21J` & `Omicron/21K`). Second, to detect recombination within Omicron (ex. `Omicron/BA.2/21L` & `Omicron/BA.5/22B`). It was not possible to define universal parameters for `sc2rf` that worked for both distantly related clades, and the closely related Omicron lineages. | ||
| - [Issue #51](https://github.com/ktmeaton/ncov-recombinant/issues/51): Rename parameter `clades` to `primary_clades` and add new parameter `secondary_clades` for detecting BA.5. | ||
| - [Issue #53](https://github.com/ktmeaton/ncov-recombinant/issues/53): Identify the parental lineages by splitting up the observed mutations (from `nextclade`) into regions by breakpoint. Then query the list of mutations in <https://cov-spectrum.org> and report the lineage with the highest prevalence. | ||
| - Tested out `--enable-deletions` again, which caused issues for `XD`. This confirms that using deletions is still ineffective for defining breakpoints. | ||
| - Add `B.1.631` and `B.1.634` to `sc2rf/mapping.tsv` and as potential clades in the default parameters. These are parents for `XB`. | ||
| - Add `B.1.438.1` to `sc2rf/mapping.tsv` and as a otential clade in the default parameters. This is a parent for [`proposed808`](https://github.com/cov-lineages/pango-designation/issues/808). | ||
| - Require a recombinant region to have at least one substitution unique to the parent (i.e. diagnostic). This reduces false positives. | ||
| - Remove the debugging mode, as it produced overly verbose output. It is more efficient to rerun manually with custom parameters tailored to the kind of debugging required. | ||
| - Change parent clade nomenclature from `Omicron/21K` to the more comprehensive `Omicron/BA.1/21K`. This makes it clear which lineage is involved, since it's not always obvious how Nextclade clades map to pango lineages. | ||
|
|
||
| ## UShER | ||
|
|
||
| - [Issue #63](https://github.com/ktmeaton/ncov-recombinant/issues/63): All UShER rules and output have been removed. First, because the latest releases of nextclade datasets (tag `2022-07-26T12:00:00Z`) have dramatically improved lineage assignment accuracy for recombinants. Second, was to improve runtime and simplicity of the workflow, as UShER adds significantly to runtime. | ||
|
|
||
| ## Linelist | ||
|
|
||
| - [Issue #30](https://github.com/ktmeaton/ncov-recombinant/issues/30): Fixed the bug where distinct recombinant lineages would occasionally be grouped into one `cluster_id`. This is due to the new definition for recombinant lineages (see General) section, which now includes parental _lineages_ and have sufficient resolving power. | ||
| - [Issue #46](https://github.com/ktmeaton/ncov-recombinant/issues/46): Added new column `parents_subs`, which are the substitutions found in the parental lineages _before_ recombination occurred using the `sars-cov-2-no-recomb` nextclade dataset. Also added new columns: `parents_lineage`, `parents_lineage_confidence`, based on querying `cov-spectrum` for the substitutions found in `parents_subs`. | ||
| - [Issue #53](https://github.com/ktmeaton/ncov-recombinant/issues/53): Added new column `cov-spectrum_query` which includes the substitutions that are shared by ALL sequences of the recombinant lineage. | ||
| - Added new column `cluster_privates` which includes the private substitutions shared by ALL sequences of the recombinant lineage. | ||
| - Renamed `parents` column to `parents_clade`, to differentiate it from the new column `parents_lineage`. | ||
|
|
||
| ## Plot | ||
|
|
||
| - [Issue #4](https://github.com/ktmeaton/ncov-recombinant/issues/4), [Issue #57](https://github.com/ktmeaton/ncov-recombinant/issues/57): Plot distributions of each parent separately, rather than stacking on one axis. Also plot the substitutions as ticks on the breakpoints figure. | ||
|
|
||
| | v0.3.0 | v0.4.0 | | ||
| | -------------------------------------------------------------------------------------------------------------------------------------- | -------------------------------------------------------------------------------------------------------------------------------------- | | ||
| | 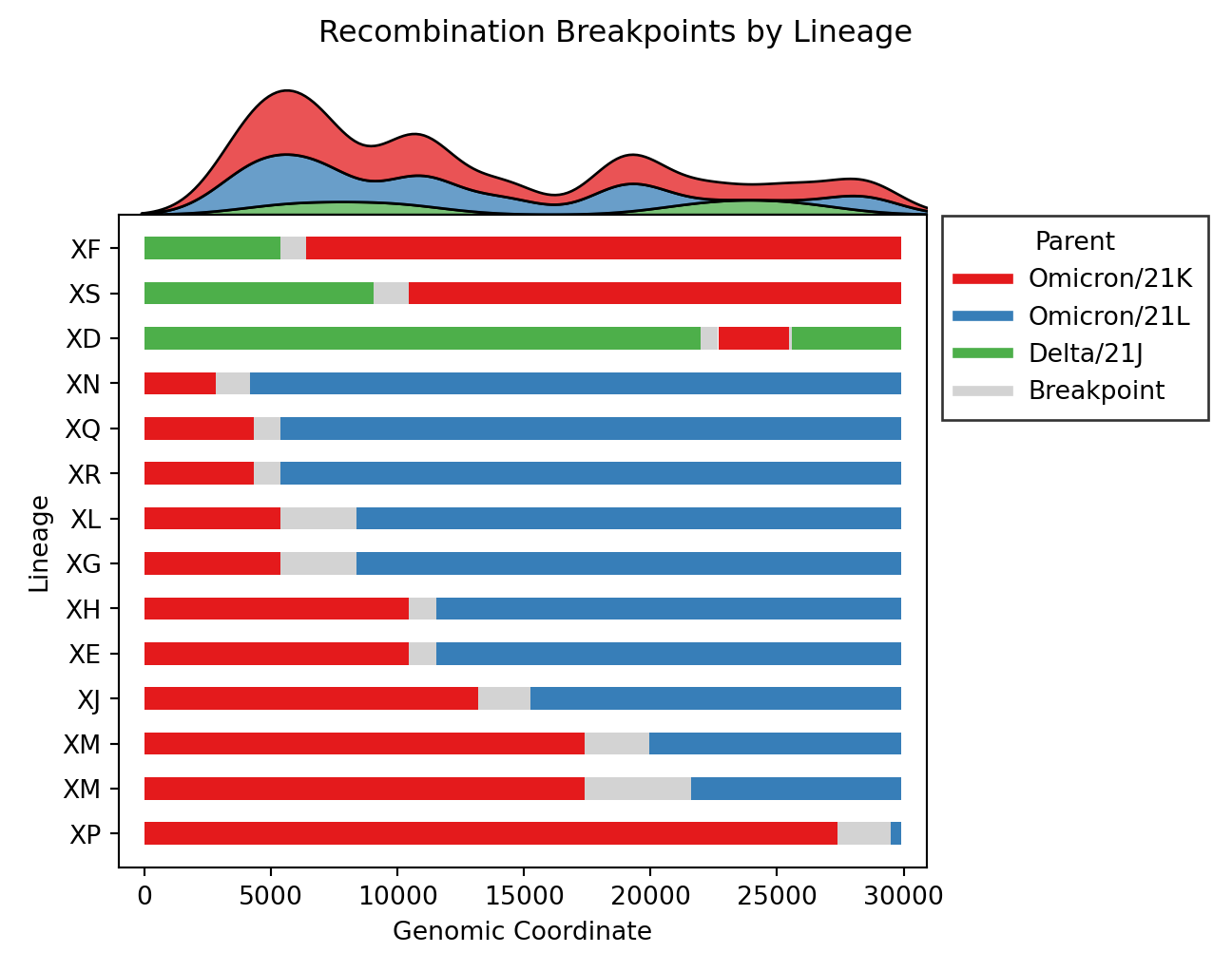 |  | | ||
|
|
||
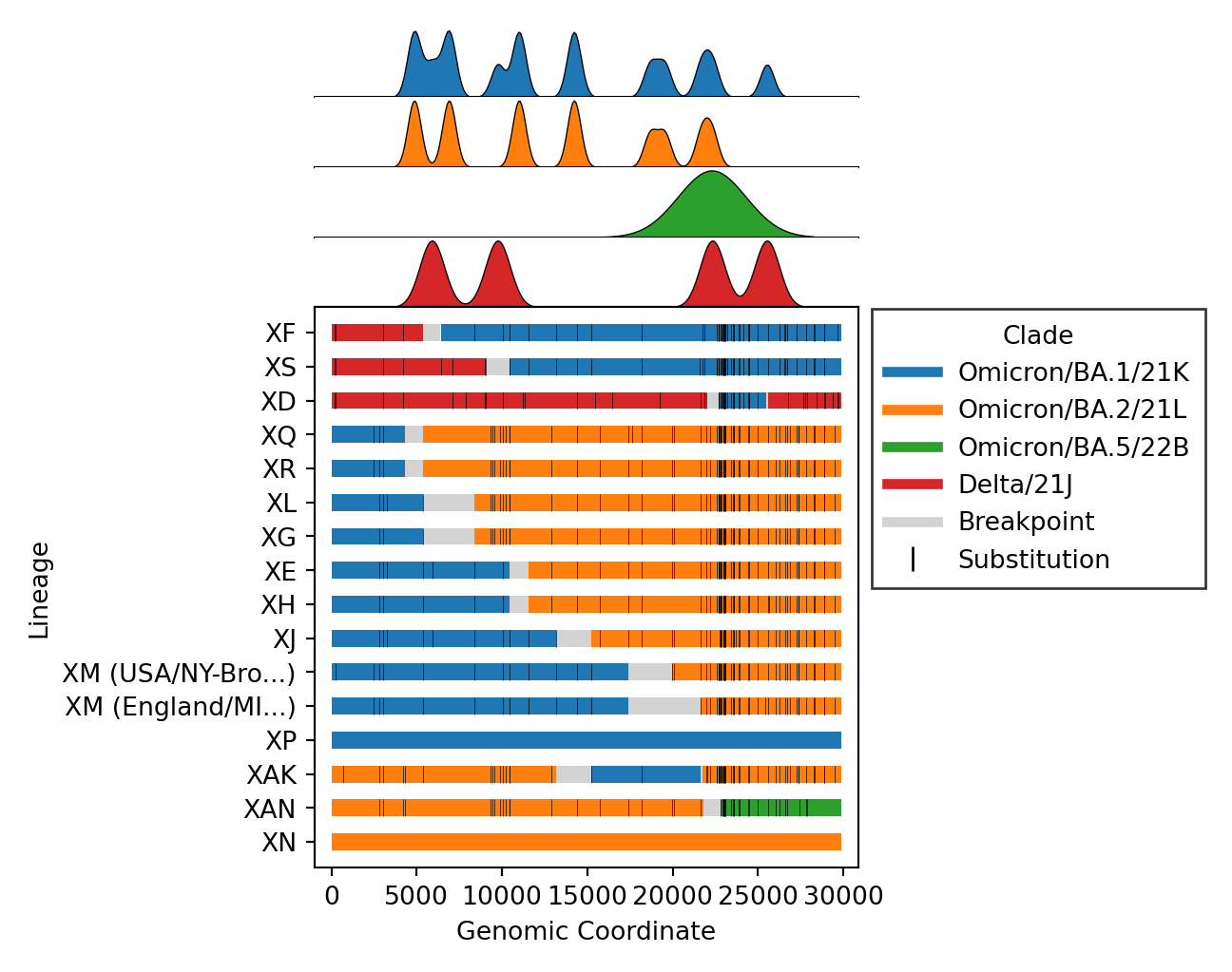
| - [Issue #46](https://github.com/ktmeaton/ncov-recombinant/issues/46): Plot breakpoints separately by clade _and_ lineage. In addition, distinct clusters within the same recombinant lineage are noted by including their cluster ID as a suffix. As an example, please see `XM (USA) and X (England)` below. Where the lineage is the same (`XM`), but the breakpoints differ, as do the parental lineages (`BA.2` vs `BA.2.12.1`). These clusters are distinct because `XM (England)` lacks substitutions occurring around position 20000. | ||
|
|
||
| | Clade | Lineage | | ||
| |:--------------------------------------------------------------------------------------------------------------------:|:-------------------------------------------------------------------------------------------------------:| | ||
| |  |  | | ||
|
|
||
| - [Issue #58](https://github.com/ktmeaton/ncov-recombinant/issues/58): Fix breakpoint plotting from all lineages to just those observed in the reporting period. Except for the breakpoint plots in `plots_historical`. | ||
| - [Issue #59](https://github.com/ktmeaton/ncov-recombinant/issues/59): Improved error handling of breakpoint plotting when a breakpoint could not be identified by `sc2rf`. This is possible if `nextclade` was the only program to detect recombination (and thus, we have no breakpoint data from `sc2rf`). | ||
| - [Issue #64](https://github.com/ktmeaton/ncov-recombinant/issues/64): Improved error handling for when the lag period (ex. 4 weeks) falls outside the range of collection dates (ex. 2 weeks). | ||
| - [Issue #65](https://github.com/ktmeaton/ncov-recombinant/issues/65): Improved error handling of distribution plotting when only one sequence is present. | ||
| - [Issue #67](https://github.com/ktmeaton/ncov-recombinant/issues/67): Plot legends are placed above the figure and are dynamically sized. | ||
|
|
||
| | v0.3.0 | v0.4.0 | | ||
| | ----------------------------------------------------------------------------------------------------------------------------------------------------------------- | ----------------------------------------------------------------------------------------------------------------------------------------------------------------- | | ||
| | 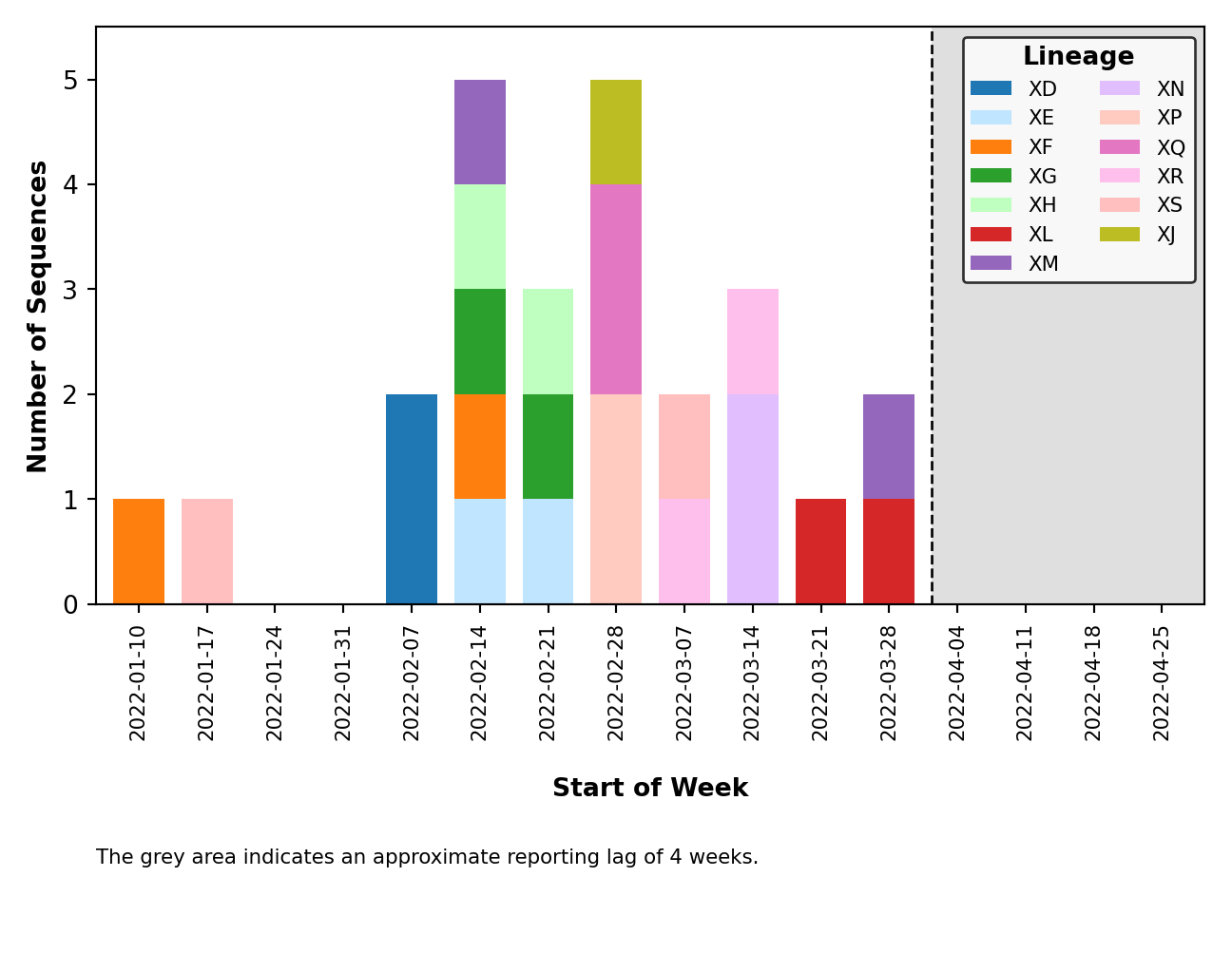 | 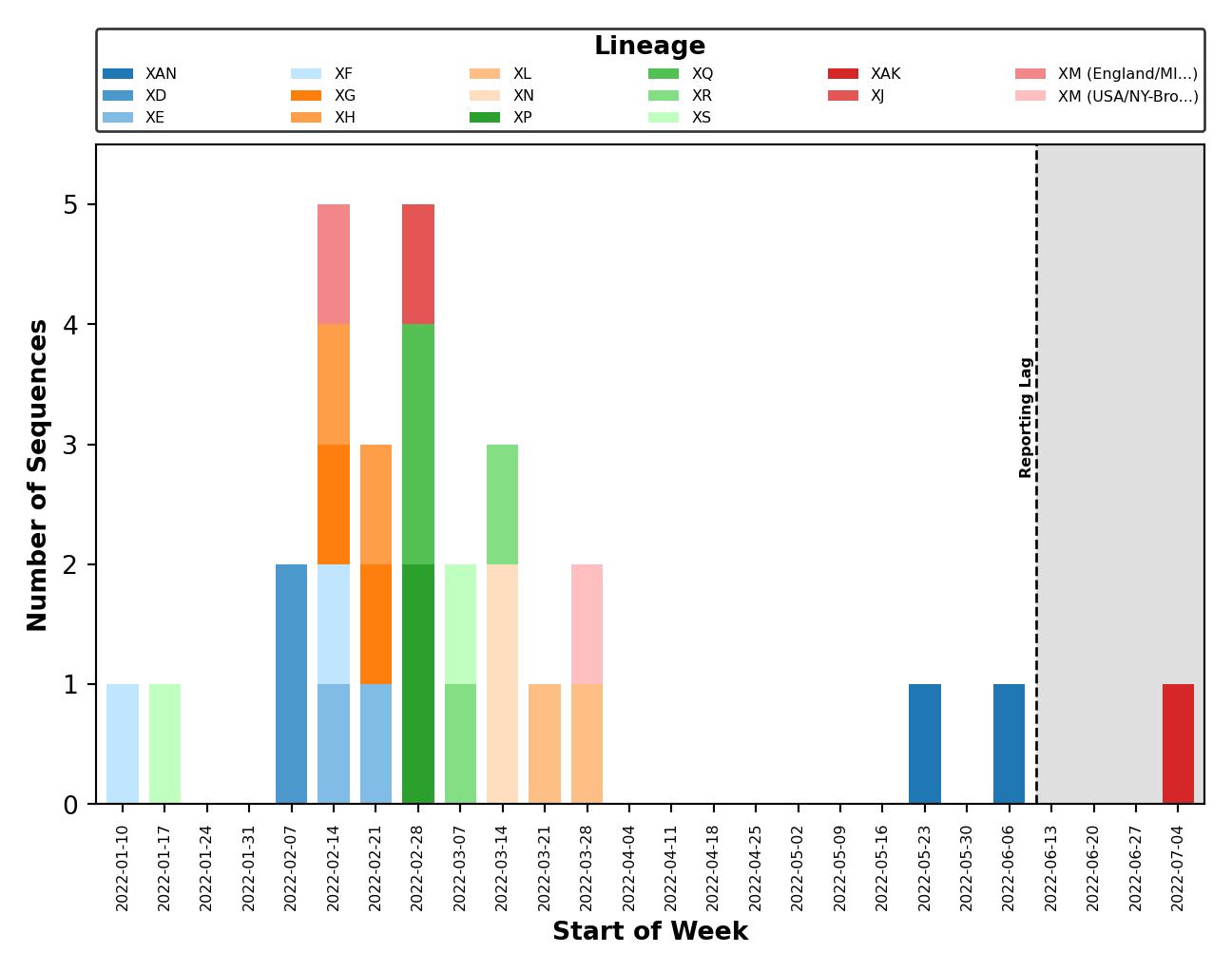 | | ||
|
|
||
| ## Report | ||
|
|
||
| - [Issue #60](https://github.com/ktmeaton/ncov-recombinant/issues/60): Remove changelog from final slide, as this content did not display correctly | ||
| - [Issue #61](https://github.com/ktmeaton/ncov-recombinant/issues/61): Fixed bug in the `report.xlsx` where the number of proposed and unpublished recombinant lineages/sequences was incorrect. | ||
|
|
||
| ## Validation | ||
|
|
||
| - [Issue #58](https://github.com/ktmeaton/ncov-recombinant/issues/58): New rule (`validate`) to validate the number of positives in controlled datasets (ex. controls, tutorials) against `defaults/validation.tsv`. If validation fails based on an incorrect number of positives, the pipeline will exit with an error. This is to make it more obvious when results have changed during Continuous Integration (CI) |