![]()
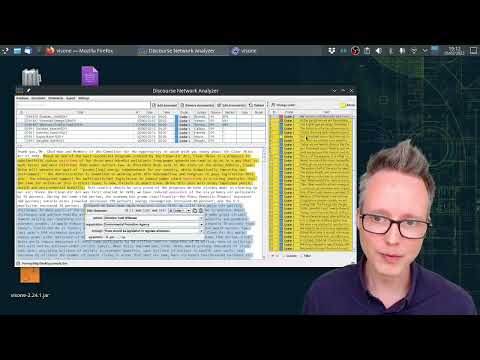
The Java software Discourse Network Analyzer (DNA) is a qualitative content analysis tool with network export facilities. You import text files and annotate statements that persons or organizations make, and the program will return network matrices of actors connected by shared concepts.
-
Download the latest release of the software.
-
Annotate documents, such as newspaper articles or speeches, with statements of what actors say; then export network data.
-
You can use the stand-alone software visone (or any other network analysis software) for analyzing the resulting networks.
-
The software comes with an R package called rDNA for remote controlling DNA and for further ways of analyzing the networks.
DNA 3.0 was first released on 12 June 2022. It constitutes a major rewrite from the previous version DNA 2.0 beta 25. DNA 3 comes with many new features and improvements. The release page contains all the details (scroll to version 3.0.7 for the first DNA 3 release).
If you require the latest (non-release) version of the DNA jar file from GitHub, you can clone the git repository to your computer and execute ./gradlew build on your terminal or command line. This will build the jar file and store it in the build/ directory of the cloned repository. Alternatively, you can try to download the latest artifact from the build process under GitHub Actions by clicking on the latest build and scrolling down to "Artifacts". However, it is usually recommended to use the most recent release version.
![]()
The R package rDNA connects DNA to R for data exchange and analysis.
Please note that the current version 3.0 does not have the full functionality of the old 2.0 version yet. It can create networks, but please use the old version for now if you require more complex data management and analysis functionality in R. It is possible to import DNA 2 data into DNA 3 at any point (but not the other way around). New R functions will be added in the future.
To install the new rDNA 3 directly from GitHub, try the following code in R:
# install.packages("remotes")
remotes::install_github("leifeld/dna/rDNA/rDNA@*release",
INSTALL_opts = "--no-multiarch")Note that the package relies on rJava, which needs to be installed first.
For data management, you may still want to use the old rDNA 2.1.18 with DNA 2.0 beta 25. You can install the package directly from GitHub as well. However, you will need to download the correct JAR file and store it either in your working directory or (recommended) in the library path of the installed R package in the "extdata" subdirectory. The following code can do this for you:
# install.packages("remotes")
remotes::install_github("leifeld/dna/rDNA@v2.0-beta.25",
INSTALL_opts = "--no-multiarch")
# find out where to store the JAR file
dest <- paste0(dirname(system.file(".", package = "rDNA")),
"/extdata/dna-2.0-beta25.jar")
# download JAR file and store in library path
u <- "https://github.com/leifeld/dna/releases/download/v2.0-beta.25/dna-2.0-beta25.jar"
download.file(url = u, destfile = dest, mode = "wb")
-
This tutorial on YouTube describes installation of DNA, basic data coding, network export, and network analysis using visone. The video clip is 18 minutes long.
-
See the bibliography for several hundred publications and theses using discourse network analysis or the DNA software.
-
The introductory chapter (Leifeld 2017) in the Oxford Handbook of Political Networks is recommended as a primer (chapter; preprint).
-
The previous version of DNA and rDNA came with a detailed manual of more than 100 pages. It is outdated, but perhaps still useful.
-
If you have questions or want to report bugs, please create an issue in the issue tracker.
-
Join the the DNA community on Matrix. Matrix is a chat protocol. It's similar to Slack, Discord, or WhatsApp, but without the corporate shackles. It's free, open-source, decentralised, and secure. We have set up a public space called #dna:yatrix.org with separate chat rooms for installation, research, and development. It's really easy to join: You first create an account on one of the many Matrix servers (we use and recommend yatrix.org), then download one of the many Matrix clients on your phone, computer, or the web (e.g., Element) to use the account with, and finally join #dna:yatrix.org. To simplify the process, you can just click on this invitation link for some sensible default choices. Make sure you join all four public rooms (you can mute their notifications as needed) and look at the rules in the #dna-welcome room upon arrival.
Please consider contributing to the project by:
- telling other people about the software,
- citing our underlying research in your publications,
- reporting or fixing issues, or
- starting pull requests to contribute bug fixes or new functionality.
Some suggestions of new functionality you could add via pull requests:
- Import filters for loading data from Nvivo, MaxQDA, and other software into DNA.
- Export filters for exporting networks to Gephi and other network analysis software.
- Analysis functions or unit tests for the rDNA package.
- Publications for the bibliography.
- Bug fixes.