{kind=link}
{kind=link}
{kind=link}
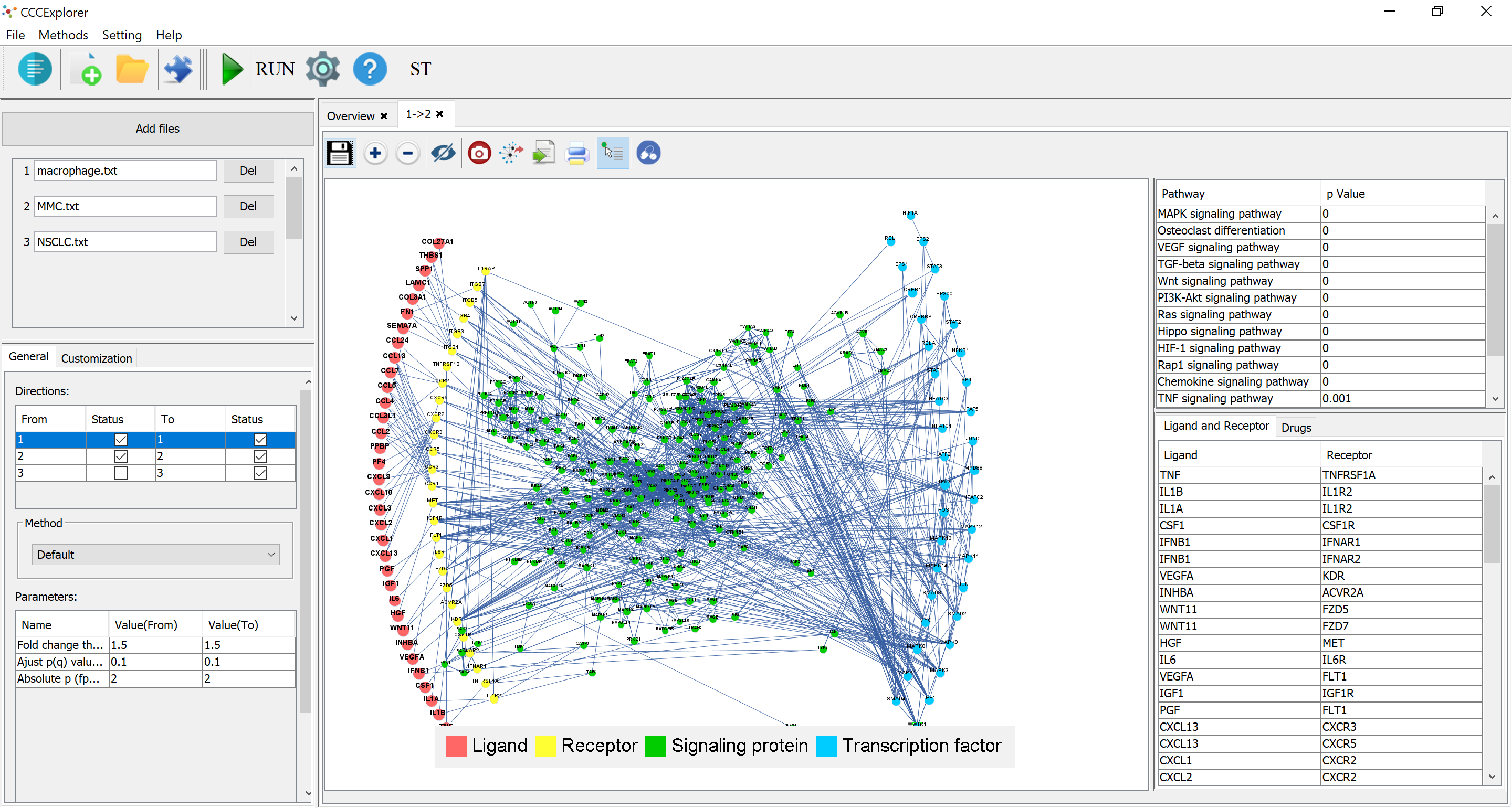
CCCExplorer is a java-based software that predicts and visualizes the gene signaling network to aid research on crosstalk identification in the tumor microenvironment. CCCExplorer integrates a computational model that we developed to uncover cell-cell communication as a direct and connected network. These cell communications range from ligand-receptor interactions to transcription factors and their target genes.
Transcriptome analysis of individual stromal cell populations identifies stroma-tumor crosstalk in mouse lung cancer model. H Choi, J Sheng, et al. Cell Reports, 2015. 24;10(7):1187-201
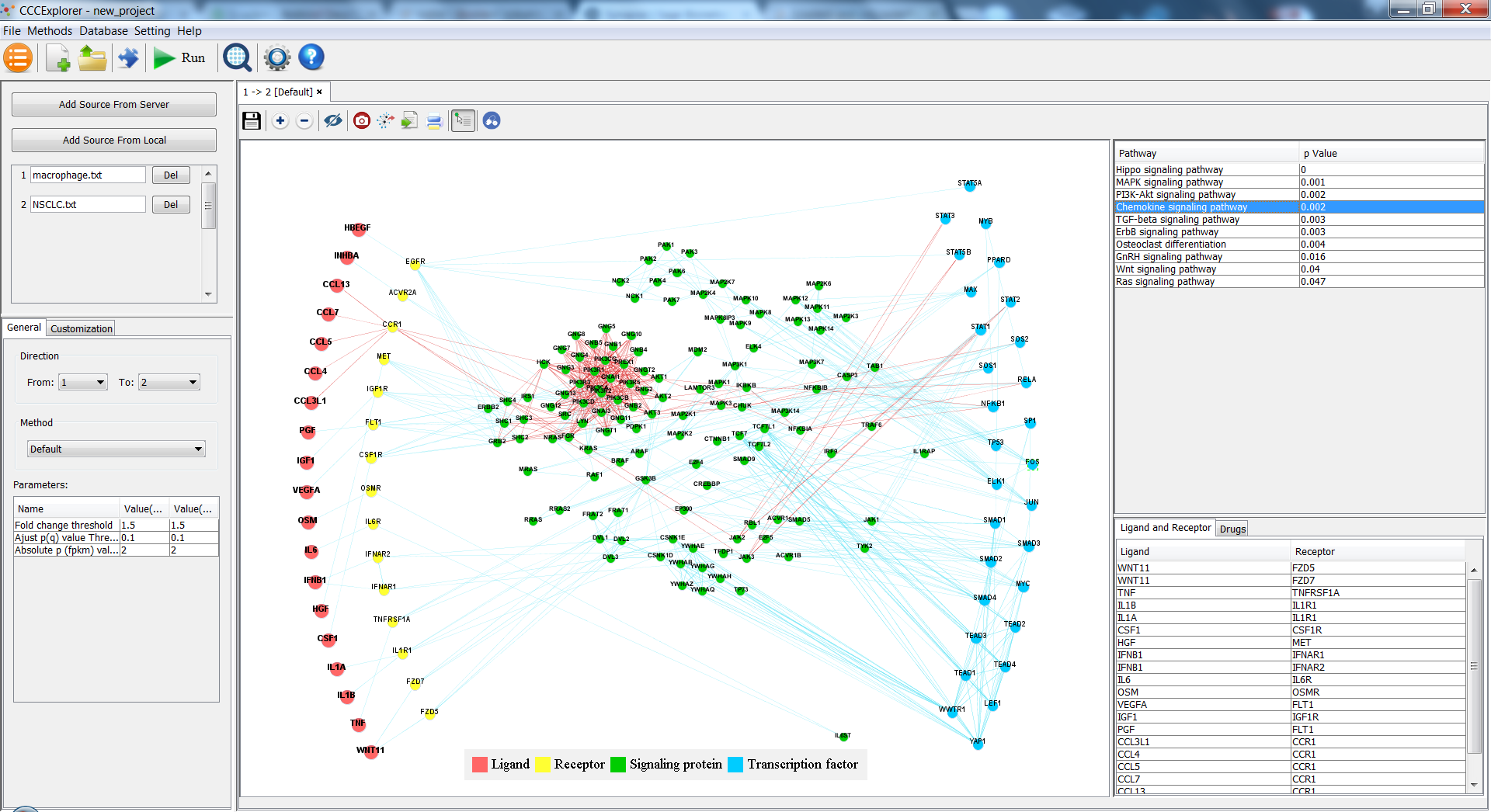
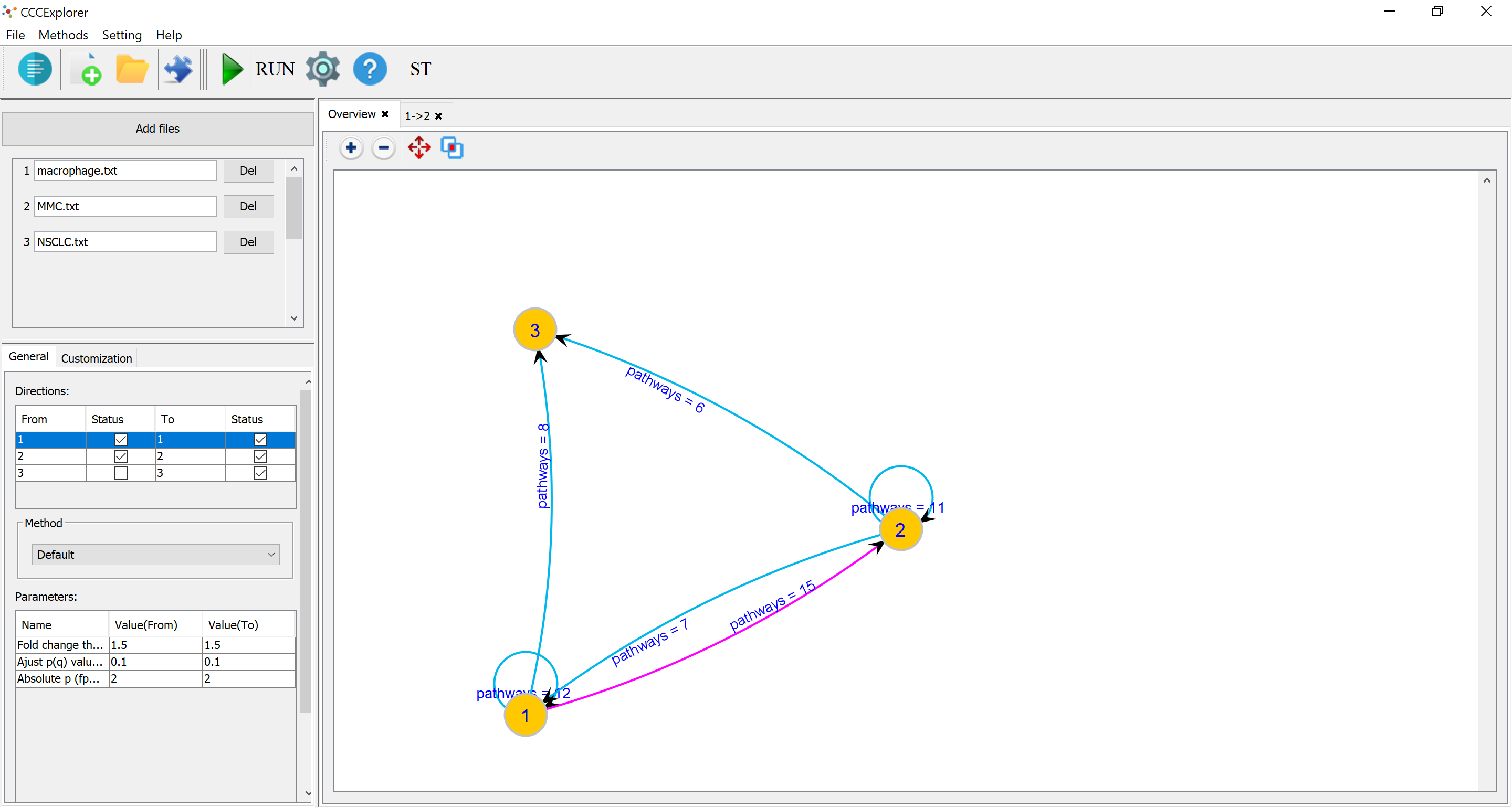
It support multiple cell types and other new features.
https://www.dropbox.com/s/1y1or8ougjkzu3x/CCCExplorer_MacOS.zip?dl=0
Youtube video URL:
Please read the guide before you run CCCExplorer
https://github.com/methodistsmab/CCCExplorer/blob/master/CCCExplorer%20user_guide_v1.pdf
https://github.com/methodistsmab/CCCExplorer/blob/master/CCCExplorer_End_User_License_Agreement.pdf
Xiaohui Yu
Email: xyu2@houstonmethodist.org
Jianting Sheng
Email: jsheng@houstonmethodist.org
Wong, Stephen (PI)
Email: STWong@houstonmethodist.org