{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

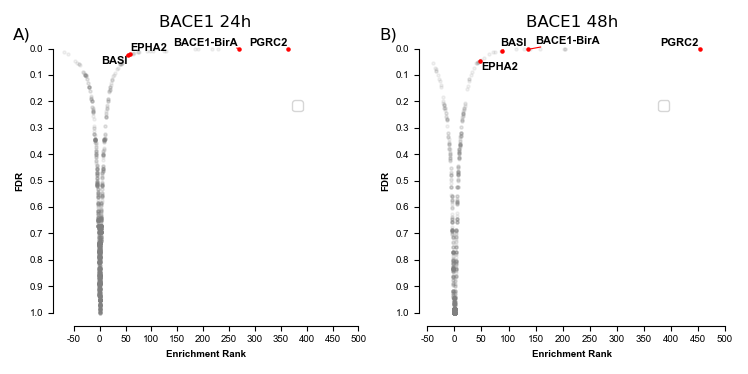
This repository contains the data analysis of the BioID proximity assay used to establish the BACE1 interactome in healthy neuronal cells.
Use the package manager conda to install the environment or start the mybinder server using this badge
conda env create -f environment.yml
conda activate myenv
jupyter notebook
## start analysis_def.ipynb