![]()
![]()
![]()
1. Make a GitHub Account
You'll need to make a GitHub Account.
2. Click Edit Button on Page You Want to Edit

3. Fork the Repository When Prompted (only the first time)
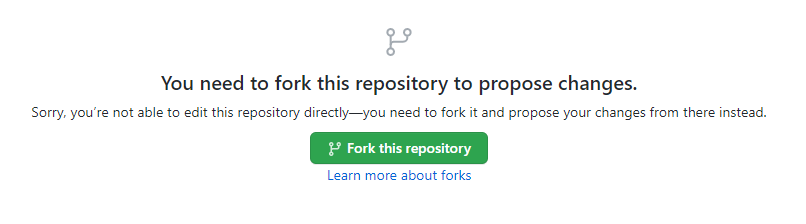
{kind=link}
4. Make the Edits in MarkDown

5. Propose Changes
Please describe the change you are making.

6. Create Pull Request

7. Finalize Pull Request with Description

Follow steps 1 - 3.
Navigate to mzmine_documentation/docs in your fork and create a new file

Follow steps 4 - 7.
{{ git_page_authors }}
This page was adapted from the GNPS documentation.