By Paul Hockett with Varun Makhija
Quantum Metrology with Photoelectrons Volume 3: Analysis methodologies, an open source executable book. This repository contains the source documents (mainly Jupyter Notebooks in Python) and notes for the book, and the current HTML build can be found online, which includes all code and interactive figures; a PDF version (with abridged code) is also available. The book is published by IOP Press (Dec. 2023), and print editions are available - see the IOP catalogue page for details.
Photoionization is an interferometric process, in which multiple paths can contribute to the final continuum photoelectron wavefunction. At the simplest level, interferences between different final angular momentum states are manifest in the energy and angle resolved photoelectron spectra: metrology schemes making use of these interferograms are thus phase-sensitive, and provide a powerful route to detailed understanding of photoionization. In these cases, the continuum wavefunction (and underlying scattering dynamics) can be characterised. At a more complex level, such measurements can also provide a powerful probe for other processes of interest, leading to a more general class of quantum metrology built on phase-sensitive photoelectron imaging. Since the turn of the century, the increasing availability of photoelectron imaging experiments, along with the increasing sophistication of experimental techniques, and the availability of computational resources for analysis and numerics, has allowed for significant developments in such photoelectron metrology.

-
Volume I covers the core physics of photoionization, including a range of computational examples. The material is presented as both reference and tutorial, and should appeal to readers of all levels. ISBN 978-1-6817-4684-5, http://iopscience.iop.org/book/978-1-6817-4684-5 (IOP Press, 2018)
-
Volume II explores applications, and the development of quantum metrology schemes based on photoelectron measurements. The material is more technical, and will appeal more to the specialist reader. ISBN 978-1-6817-4688-3, http://iopscience.iop.org/book/978-1-6817-4688-3 (IOP Press, 2018)
Additional online resources for Vols. I & II can be found on OSF and Github.
- Volume III in the series continues this exploration, with a focus on numerical analysis techniques, forging a closer link between experimental and theoretical results, and making the methodologies discussed directly accessible via new software. The book is due for publication by IOP in 2023; this volume is also open-source, with a live HTML version at https://phockett.github.io/Quantum-Metrology-with-Photoelectrons-Vol3/ and source available at https://github.com/phockett/Quantum-Metrology-with-Photoelectrons-Vol3.
This repository contains:
doc-sourcethe source documents (mainly Jupyter Notebooks in Python)notesadditional notes for the book,- gh-pages branch contains the current HTML build, available at https://phockett.github.io/Quantum-Metrology-with-Photoelectrons-Vol3/
The project has been setup to use the Jupyter Book build-chain (which uses Sphinx on the back-end) to generate HTML and Latex outputs for publication from source Jupyter notebooks & markdown files.
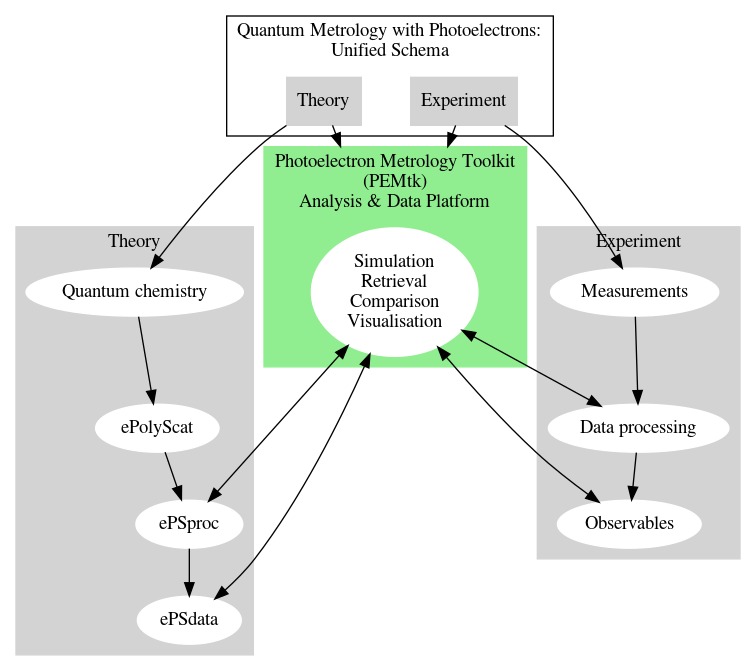
The work within the book will make use of the Photoelectron Metrology Toolkit platform for working with experimental & theoretical data.
For further technical details, see Chpt. 2 in the book.
Each Jupyter notebook (*.ipynb) can be treated as a stand-alone computational document. These can be run/used/modified independently with an appropriately setup python environment, for details see Chpt. 2 in the book.
Docker images, including the full book source and all required packages, are available from Docker hub, simply run docker pull epsproc/quantum-met-vol3 to pull a copy, then docker run epsproc/quantum-met-vol3 to run with default settings (which uses port 8888 for JupyterLab). The Jupyter Lab interface will be available at http://localhost:8888, with default password qm3. (To specify a port at run time, add -p <newPort>:8888 to the run command, e.g. docker run -p 9999:8888 epsproc/quantum-met-vol3 to set port to 9999.)
The Docker images contain the book source, along with all required packages and Jupyter Lab (based on the Jupyter Docker Stacks Scipy image). Book source files are available in the container at github/Quantum-Metrology-with-Photoelectrons-Vol3/. For more details on the Jupyter Lab base container, see the Jupyter Docker Stacks website.
For the source Dockerfiles and additional notes, see /docker in the QM3 github repository.
Archived versions can also be found on Zenodo:
The full book can also be built from source in a suitably configured environment (see Chpt. 2 in the book):
- Clone this repository
- Run
pip install -r requirements.txt(it is recommended you do this within a virtual environment) - (Optional) Edit the books source files located in the
doc-source/directory - Run
jupyter-book clean doc-source/to remove any existing builds - Run
jupyter-book build doc-source/
A fully-rendered HTML version of the book will be built in doc-source/_build/html/.
This project is created using the open source Jupyter Book project and the executablebooks/cookiecutter-jupyter-book template.
For a full build environment listing, see Build versions.