Issue with inversion/retention of stereochemistry #2891
Comments
|
Thanks for that detailed and well described bug report! There's definitely something odd/unexpected going on here that I'm going to have to dig into (it's not a trivial topic) in order to understand/explain. |
greglandrum
added a commit
to greglandrum/rdkit
that referenced
this issue
Sep 17, 2020
Try to be more robust w.r.t. atom reordering in input SMILES
greglandrum
added a commit
that referenced
this issue
Sep 18, 2020
* add tests for the problem * more testing (still no fix) * Fixes #2891 Try to be more robust w.r.t. atom reordering in input SMILES * better handling of differing numbers of bonds between reactants and products all tests now passing * update the rdkit book with more details about chirality in reactions * changes in response to review
|
@mc-robinson : I believe we have this fixed on master now. It'll be in the 2020.09.1 release (coming in mid-October). There's a gist with more here: |
|
Thanks @greglandrum! |
Sign up for free
to join this conversation on GitHub.
Already have an account?
Sign in to comment
Description:
Ran into a few strange behaviors (at least relative to what I would expect) when trying to code up a substitution reaction. Minimal examples reproduced below:
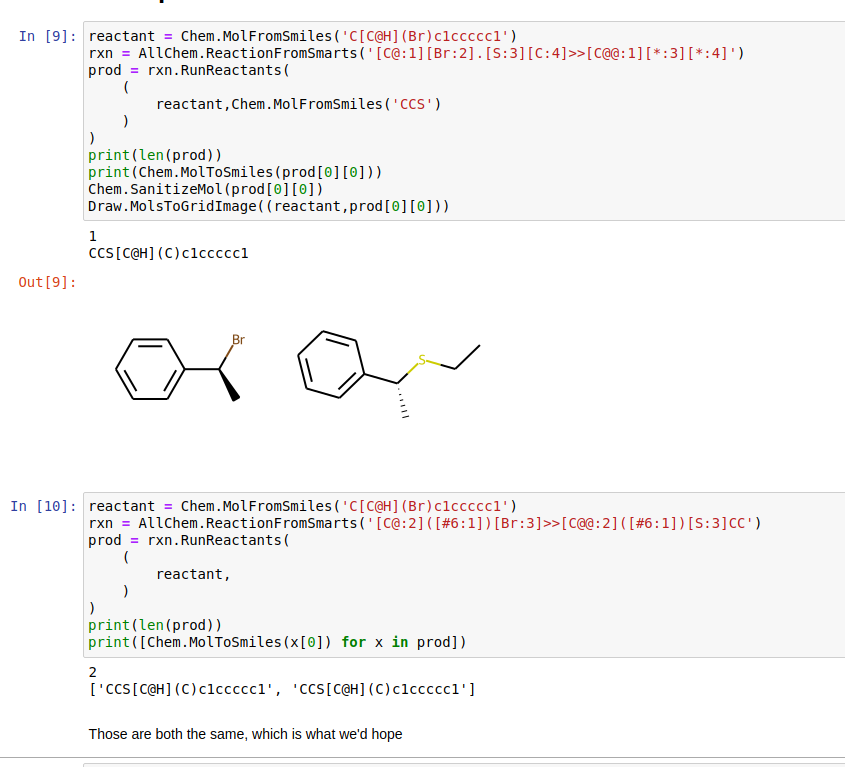
For all examples the reactant of interest is
Chem.MolFromSmiles('C[C@H](Br)c1ccccc1')We begin by testing the simple bimolecular substitution reaction, with inversion of stereochemistry at the reaction center:
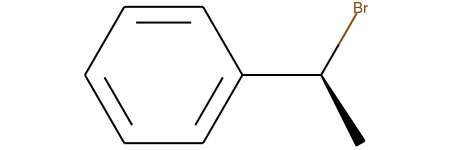
The result is
'CCS[C@@H](C)c1ccccc1'(shown below)However, this is actually not the expected inverted product. The reason for this I believe is due to the change of the order of the SMILES. The carbon indeed changes from [C@] to [C@@]; however, since the canonicalization changes the bond we look down to define clockwise/counter-clockwise, no true inversion occurs. Ultimately, I did not see an easy way to fix this problem (though I welcome suggestions!), so started testing a unimolecular transformation, as shown below.
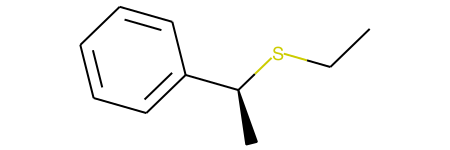
This example, produces both stereoisomers:
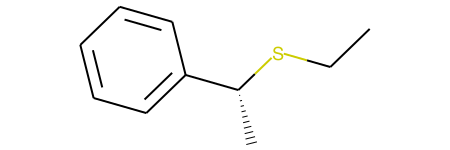
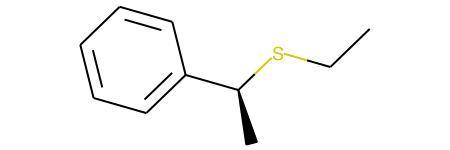
and
I understand that my RXN SMARTS is likely overly general, since
'[C@:2]([#6:1])[Br:3]'may be matched in two different ways to the reactant. However, either way it matches, shouldn't the product simply invert the stereochemistry of the original match? It is a bit unclear to me why the configuration of the reactant is retained.Investigating this behavior, I tried the less general rxn below
This actually only produces the expected product with inversion of stereochemistry:
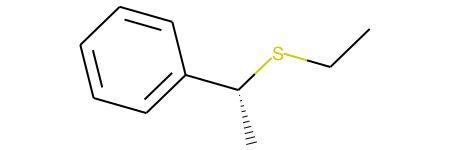
Lastly, I tried to isolate the behavior of the aromatic carbon causing troubles:
This reaction only produces the unexpected product with retention.
I expect that an issue may arise because the original SMILES of the reactant,
''C[C@H](Br)c1ccccc1'is defining counter-clockwise with respect to looking down the non-aromatic CC bond, and not the cC bond.However, I am pretty confused in general, so would really appreciate any clarification. I apologize for the lengthy issue, but hopefully the reproducible examples are of help.
The text was updated successfully, but these errors were encountered: