This is is the lora pipeline from the Sequana project
| Overview: | Run assembler (Canu, flye, hifiasm) on a set of long read files |
|---|---|
| Input: | A set of BAM files from Pacbio sequencers, or FastQ files for Nanopore sequencers. |
| Output: | HTML reports with assemblies for each sample. |
| Status: | prod |
| Citation: | Cokelaer et al, (2017), ‘Sequana’: a Set of Snakemake NGS pipelines, Journal of Open Source Software, 2(16), 352, JOSS DOI doi:10.21105/joss.00352 |
Install Lora with pip command:
pip install sequana-lora
To update your installed version, type:
pip install sequana-lora --upgrade
sequana_lora --help sequana_lora --input-directory DATAPATH
This creates a directory with the pipeline and configuration file. You will then need to execute the pipeline:
cd lora sh lora.sh # for a local run
This launch a snakemake pipeline. If you are familiar with snakemake, you can retrieve the pipeline itself and its configuration files and then execute the pipeline yourself with specific parameters:
snakemake -s lora.rules --cores 4 --stats stats.txt
Or use sequanix interface.
sequana_lora --input-directory . --pacbio cd lora
Do you need to build CCS ?
Look at the config file and the CCS section. Check that the parameters are as expected. If you wish to build so-called HiFi reads, set the min-passes to 10 and min-rq to 0.99.
do you have a blast DB
You may also edit the config file to set blast to true (you must handle the blast databases yourself)
Do you need an annotation from your contigs?
Set prokka to True (for bacterial annotation)
Want to check the core genome?
You may set busco to true to detect the core genome (you must provide a path to a valid lineage).
This pipelines requires the following executable(s):
- canu
- hifiasm
- flye
- blastn
- busco
- bwa
- ccs
- circlator
- checkm
- medaka
- minimap2
- pbindex
- polypolish
- prokka
- samtools
- sequana
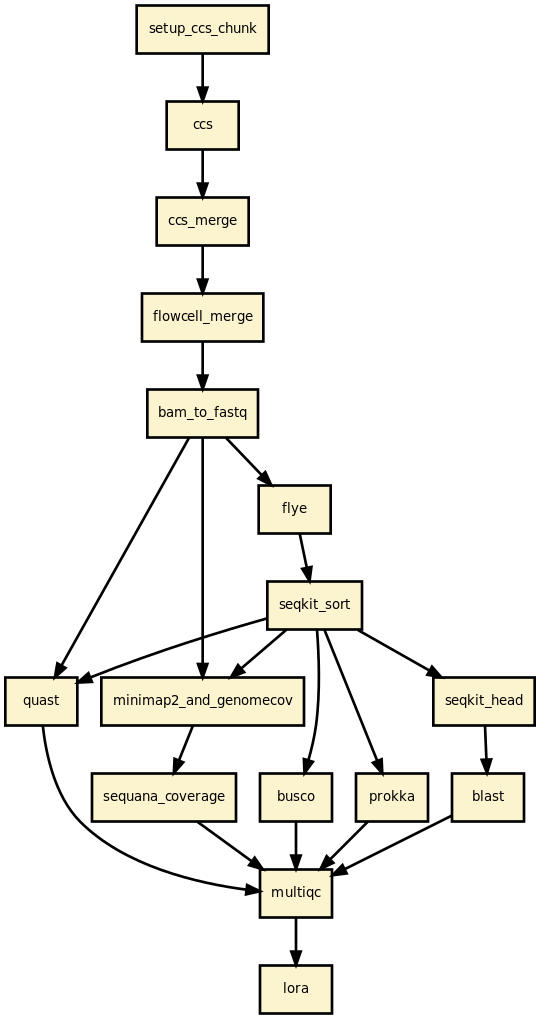
This pipeline runs lora in parallel on the input fastq files (paired or not). A brief sequana summary report is also produced.
In practice, you may start from BAM files generated by Pacbio sequencers or Fastq files, or CCS files. CCS files can be built by the pipeline. Then, an assembler is used to build the draft assemblies (Canu, hifiasm, etc). From the draft, circularisation may be applied to generate circularised genome (useful for bacterial genomes). Finally, each contig is blasted and quality checks are performed using Busco, quast, etc.
Here is the latest documented configuration file to be used with the pipeline. Each rule used in the pipeline may have a section in the configuration file.
| Version | Description |
|---|---|
| 0.3.0 |
|
| 0.2.0 |
|
| 0.1.0 | First release. |