
This is is the pacbio_qc pipeline from the Sequana projet
| Overview: | Quality control for pacbio datafiles (raw data or CCS files). |
|---|---|
| Input: | BAM files provided by Pacbio Sequencers. |
| Output: | HTML reports with various plots including taxonomic plot |
| Status: | production |
| Documentation: | This README file, the Wiki from the github repository (link above) and https://sequana.readthedocs.io |
| Citation: | Cokelaer et al, (2017), ‘Sequana’: a Set of Snakemake NGS pipelines, Journal of Open Source Software, 2(16), 352, JOSS DOI doi:10.21105/joss.00352 |
Just install this package:
pip install sequana_pacbio_qc
You will need samtools and kraken2 (optional) for the taxonomic analysis.
sequana_pacbio_qc --help sequana_pacbio_qc --input-directory DATAPATH
If you want to filter out some BAM files, you may use the pattern in tab 'input data'.
In the configuration tab, in the kraken section add as many databases as you wish. You may simply unset the first database to skip the taxonomy, which is experimental.
This creates a directory with the pipeline and configuration file. You will then need to execute the pipeline:
cd pacbio_qc sh pacbio_qc.sh # for a local run
This launch a snakemake pipeline. If you are familiar with snakemake, you can retrieve the pipeline itself and its configuration files and then execute the pipeline yourself with specific parameters:
snakemake -s pacbio_qc.rules -c config.yaml --cores 4 --stats stats.txt
Or use sequanix interface.
This pipelines requires the following executable(s):
- sequana
- samtools
- kraken2
- multiqc
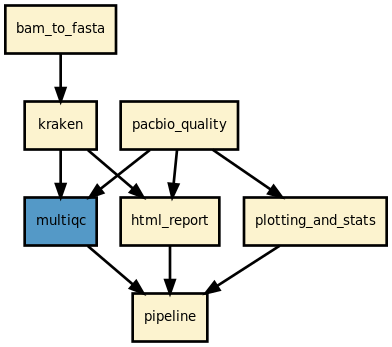
This pipeline takes as inputs a set of BAM files from Pacbio sequencers. It computes a set of basic statistics related to the read lengths. It also shows some histograms related to the GC content, SNR of the diodes and the number of passes Finally, a quick taxonomy can be performed using Kraken. HTML reports are created for each sample as well as a multiqc summary page.
Kraken databases are not provided with the pipeline. This step is optional and not used by default.
| Version | Description |
|---|---|
| 1.0.1 | fix missing import in the summary |
| 1.0.0 | Uses latest wrappers and graphviz apptainers |
| 0.11.0 | Release to use latests sequana_pipetools framework |
| 0.10.0 | Update to use latest tools from sequana framework |
| 0.9.0 | First release of sequana_pacbio_qc using latest sequana rules and modules (0.9.5) |
To contribute to this project, please take a look at the Contributing Guidelines first. Please note that this project is released with a Code of Conduct. By contributing to this project, you agree to abide by its terms.
Here is the latest documented configuration file to be used with the pipeline. Each rule used in the pipeline may have a section in the configuration file.