slaMEM is a tool used to efficiently retrieve MEMs (Maximal Exact Matches)
between a reference genome sequence and one or more query sequences, similarly to
these software tools:
slaMEM relies on an FM-Index together with a new data structure called
SSILCP (Sampled Search Intervals from Longest Common Prefixes) to store
information about parent intervals in a time- and space-efficient way.
slaMEM also includes an useful feature to display the locations of the found MEMs, generating images like the one below.
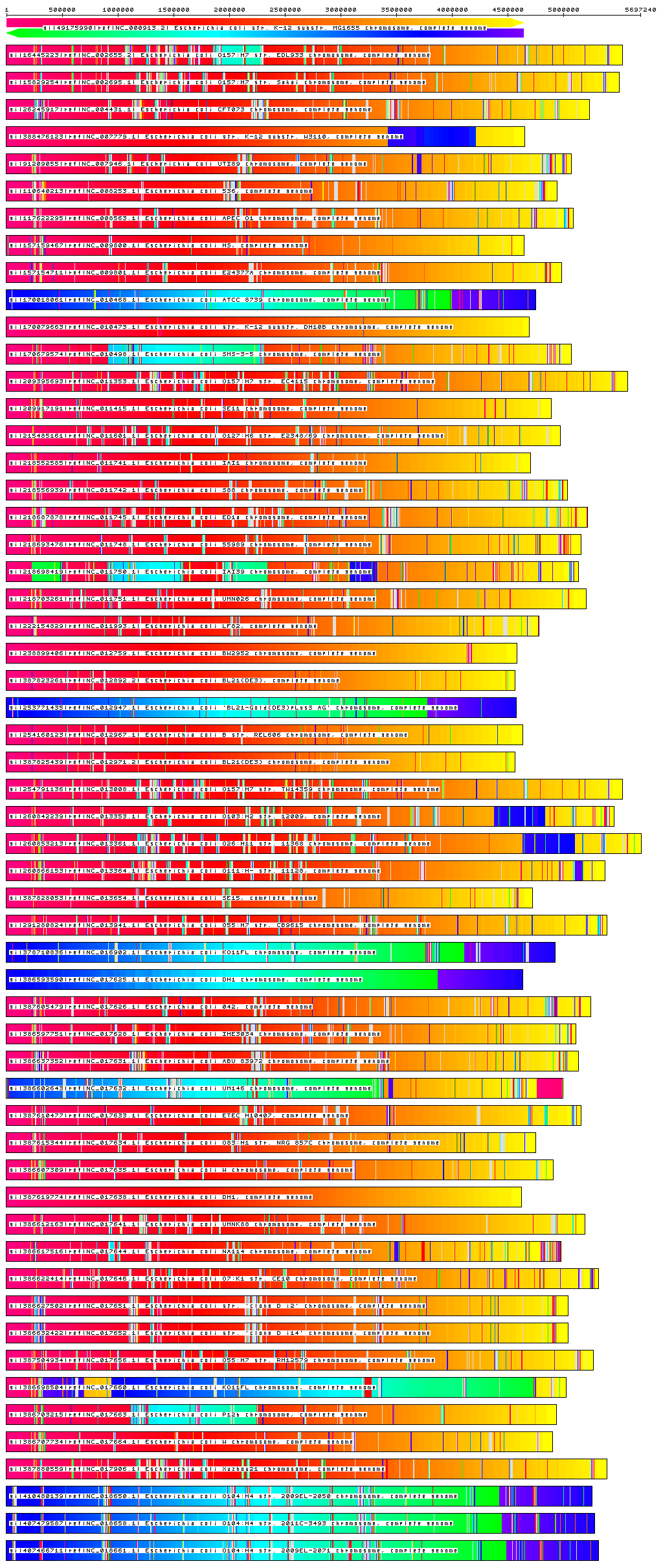
If you use slaMEM, please cite:
make./slaMEM (<options>) <reference_file> <query_file(s)>mem: find MEMs: any number of occurrences in both ref and query (default)mam: find MAMs: unique in ref but any number in queryl: minimum match length (default=20)o: output file name (default="*-mems.txt")b: process both forward and reverse strandsn: discard 'N' characters in the sequencesm: minimum sequence size (e.g. to ignore small scaffolds)r: load only the reference(s) whose name(s) contain(s) this string
v: generate MEMs map image from this MEMs file
./slaMEM -b -l 10 ./ref.fna ./query.fna
./slaMEM -v ./ref-mems.txt ./ref.fna ./query.fna