Unable to fit model #82
Comments
|
Have you computed atomrefs from QM at the level of theory that you are
using for the data?
…On Thu, May 5, 2022, 21:53 Peter Eastman ***@***.***> wrote:
I've been trying to train a model on an early subset of the SPICE dataset.
All my efforts so far have been unsuccessful. I must be doing something
wrong, but I really don't know what. I'm hoping someone else can spot the
problem. My configuration file is given below. Here's the HDF5 file for
the dataset
<https://www.dropbox.com/s/brxao096sl5poxz/SPICE-corrected.hdf5.gz?dl=0>.
I've tried training with or without derivatives. I've tried a range of
initial learning rates, with or without warmup. I've tried varying model
parameters. I've tried restricting it to only molecules with no formal
charges. Nothing makes any difference. In all cases, the loss starts out at
about 2e7 and never decreases.
The dataset consists of all SPICE calculations that had been completed
when I started working on this a couple of weeks ago. I converted the units
so positions are in Angstroms and energies in kJ/mol. I also subtracted off
per-atom energies. Atom types are the union of element and formal charge.
Here's the mapping:
typeDict = {('Br', -1): 0, ('Br', 0): 1, ('C', -1): 2, ('C', 0): 3, ('C', 1): 4, ('Ca', 2): 5, ('Cl', -1): 6,
('Cl', 0): 7, ('F', -1): 8, ('F', 0): 9, ('H', 0): 10, ('I', -1): 11, ('I', 0): 12, ('K', 1): 13,
('Li', 1): 14, ('Mg', 2): 15, ('N', -1): 16, ('N', 0): 17, ('N', 1): 18, ('Na', 1): 19, ('O', -1): 20,
('O', 0): 21, ('O', 1): 22, ('P', 0): 23, ('P', 1): 24, ('S', -1): 25, ('S', 0): 26, ('S', 1): 27}
If anyone can provide insight, I'll be very grateful!
activation: silu
atom_filter: -1
batch_size: 128
cutoff_lower: 0.0
cutoff_upper: 8.0
dataset: HDF5
dataset_root: SPICE-corrected.hdf5
derivative: false
distributed_backend: ddp
early_stopping_patience: 40
embedding_dimension: 64
energy_weight: 1.0
force_weight: 0.001
inference_batch_size: 128
lr: 1.e-4
lr_factor: 0.8
lr_min: 1.e-7
lr_patience: 10
lr_warmup_steps: 5000
max_num_neighbors: 80
max_z: 28
model: equivariant-transformer
neighbor_embedding: true
ngpus: -1
num_epochs: 1000
num_heads: 8
num_layers: 5
num_nodes: 1
num_rbf: 64
num_workers: 4
rbf_type: expnorm
save_interval: 5
seed: 1
test_interval: 10
test_size: 0.01
trainable_rbf: true
val_size: 0.05
weight_decay: 0.0
—
Reply to this email directly, view it on GitHub
<#82>, or unsubscribe
<https://github.com/notifications/unsubscribe-auth/AB3KUOWZTJQVV5OPWE5TB4DVIQRNDANCNFSM5VGDF2CA>
.
You are receiving this because you are subscribed to this thread.Message
ID: ***@***.***>
|
|
I obtained them by doing a least squares fit to the full dataset to find per-atom-type energies that best matched the total energy of every sample. |
|
I'm not able to download the dataset. When I click on download nothing happens. I tried different browsers and downloading from a terminal but I just get a connection timeout. Is it possible to get it somewhere else? |
|
I just tested and it worked fine for me. It's just on Dropbox. I would attach it here, but it's about 360 MB. Any suggestions for other places to post it? |
|
Now it works for me too. Didn't change anything... I'll try to see if I can figure something out. |
|
I'm having a couple of problems with getting it to train without errors. Have you made modifications to the code to handle the formal charge? Most of the atom types are fine but I get a few molecules where the atom types just seem broken. Then, if I just skip the atoms with atom types outside [0, 27] (the I increased I computed the mean and standard deviation of the energy on a subset (first 8000 molecules) of the dataset and got a mean around -70 and standard deviation of ~740. This doesn't really seem reasonable to me, I don't think the model will work well, if at all, if the distribution of energy is so wide. These are some random energies from the dataset: The model won't learn anything efficiently with this. I also computed mean and standard deviation for the last ~60000 molecules in the dataset and got mean -5e13 and standard deviation 1e16. It seems to me like some of the data is broken. Edit:
|
|
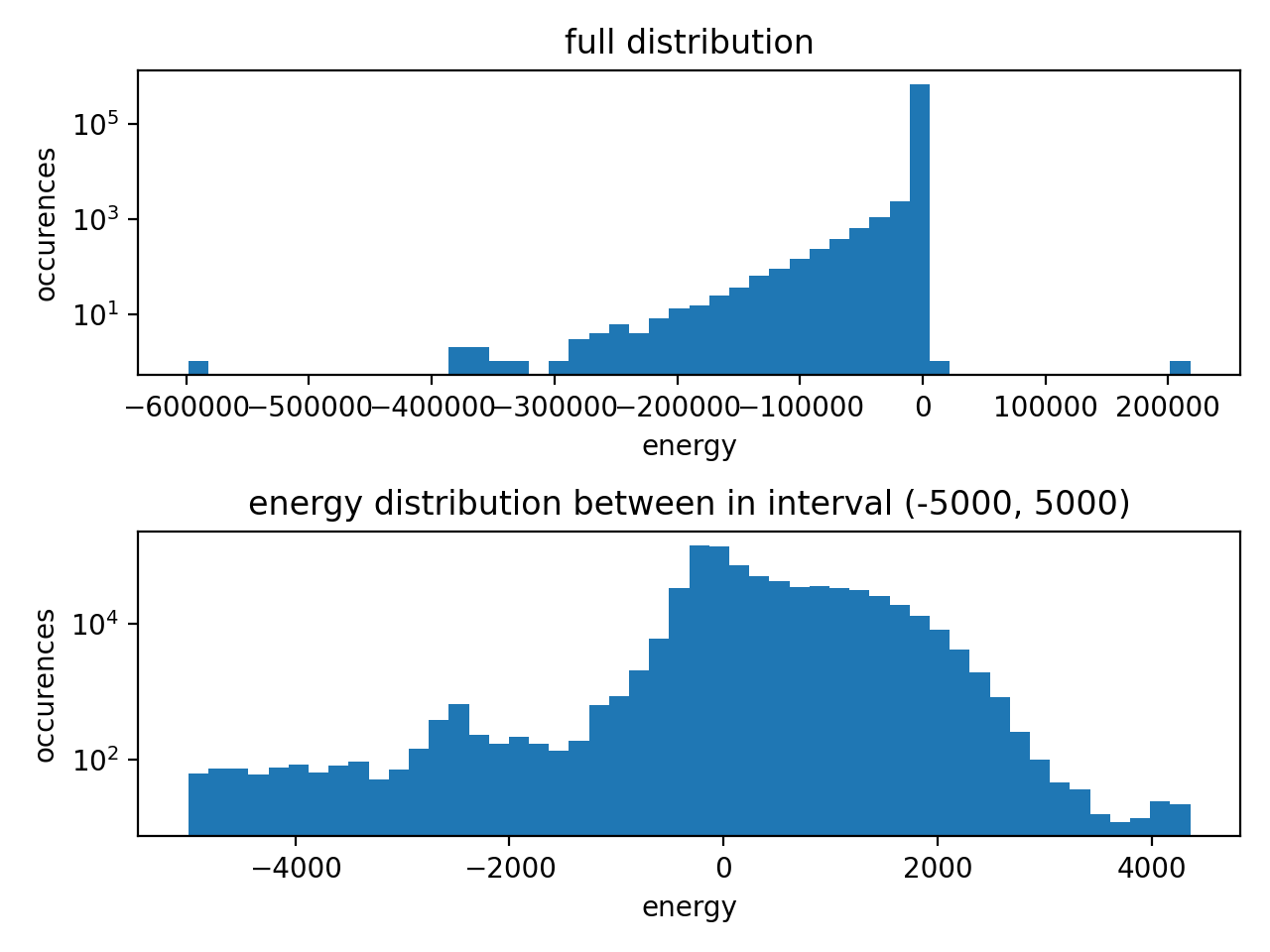
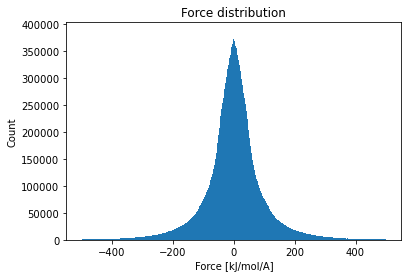
A while ago I tried to compute SPICE with xTB, but wasn't able to train. One issue was the number of neighbors (#81), but probably there are more. I tried to analyze the dataset:
|


|
My recommendation is to filter out the samples with large forces and underrepresented types. If it doesn't work, there still might be some issues with TorchMD-NET. |
I just loaded the dataset with the |
|
I just ran this test to check for negative types: f = h5py.File('SPICE-corrected.hdf5')
for g in f:
if np.any(np.array(f[g]['types']) < 0):
print('error')It didn't find anything. Where exactly do you see them? |
|
I'll try excluding the 1% of samples with the highest and lowest energies and see if that helps. Note that some of the variation in energy comes from differences in size. The samples range from 2 atoms up to nearly 100. Since the model predicts per-atom energies, that shouldn't be a problem for it. Here's a plot of the central 98% of energies.
And here's the same plot, but showing energy per atom instead of total energy.
|


|
The negative atom types are very peculiar. I can catch negative atom types with a debugger and I consistenly get broken atom types for the current index. When I retrieve another index, followed by the seemingly broken index again, I get normal looking atom types. I guess this breaks some caching that h5py might be doing. I also only get broken atom types with
I believe pytorch-lightning opens the dataset once and then copies it to the different worker processes, which is not recommended by h5py. My guess is that data gets corrupted there, which I guess could also mess with your data, although I'm confused by me being the only one where it fully breaks. It might be worth storing the dataset in something other than HDF5 or loading the full dataset into memory before training, using only a single process. When I set |
|
It must be opened in the get function the first time unless it's loaded
into memory in _init and then never access again.
…On Fri, May 6, 2022, 18:57 Philipp Thölke ***@***.***> wrote:
The negative atom types are very peculiar. I can catch negative atom types
with a debugger and when I consistenly get broken atom types for the
current index. When I retrieve another index, followed by the seemingly
broken index again, I get normal looking atom types. I guess this breaks
some caching that h5py might be doing. I also only get broken atom types
with num_workers > 1, which makes me think that the problem has to do
with multiprocessing. The h5py documentation states
Read-only parallel access to HDF5 files works with no special preparation:
each process should open the file independently and read data normally
(avoid opening the file and then forking).
I believe pytorch-lightning opens the dataset once and then copies it to
the different worker processes, which is not recommended by h5py. My guess
is that data gets corrupted there, which I guess could also mess with your
data, although I'm confused by me being the only one where it fully breaks.
It might be worth storing the dataset in something other than HDF5 or
loading the full dataset into memory before training, using only a single
process. When I set num_workers to 1 for testing my loss also seems to
decrease with only some batches spiking to 10^7 which I would assume comes
from outliers.
—
Reply to this email directly, view it on GitHub
<#82 (comment)>,
or unsubscribe
<https://github.com/notifications/unsubscribe-auth/AB3KUOVGQWEP76OB273S2ETVIVFNZANCNFSM5VGDF2CA>
.
You are receiving this because you commented.Message ID:
***@***.***>
|
|
I'll try converting everything to numpy arrays in memory and see if that makes a difference. It's not an option for really large datasets, of course, but this one is small enough it shouldn't be a problem. For the moment, though, excluding the energy outliers seems to be helping. The loss is lower, and I'm seeing a slow but steady improvement with training. My tentative conclusion is that was the main problem. I'll continue to experiment. |
|
A while ago @raimis created some benchmarks for different ways of loading large datasets. I believe memory mapped numpy arrays were the best option he found but I'm not 100% sure. |
|
yes, copy the new ANI1 dataloader in torchmd-net
…On Fri, May 6, 2022 at 7:49 PM Philipp Thölke ***@***.***> wrote:
A while ago @raimis <https://github.com/raimis> created some benchmarks
for different ways of loading large datasets. I believe memory mapped numpy
arrays were the best option he found but I'm not 100% sure.
—
Reply to this email directly, view it on GitHub
<#82 (comment)>,
or unsubscribe
<https://github.com/notifications/unsubscribe-auth/AB3KUOQGNVLL26P4SXAIBXLVIVLSXANCNFSM5VGDF2CA>
.
You are receiving this because you commented.Message ID:
***@***.***>
|
|
If I omit the samples with extreme energies and forces, training works as expected. I'm happy to close this issue, although maybe you want to keep it open to track the problem with the atom types? |
|
Can you make a PR with the spice data loader?
…On Wed, May 11, 2022, 20:28 Peter Eastman ***@***.***> wrote:
If I omit the samples with extreme energies and forces, training works as
expected. I'm happy to close this issue, although maybe you want to keep it
open to track the problem with the atom types?
—
Reply to this email directly, view it on GitHub
<#82 (comment)>,
or unsubscribe
<https://github.com/notifications/unsubscribe-auth/AB3KUOVKO2TSXOYIP4LUCJLVJP33XANCNFSM5VGDF2CA>
.
You are receiving this because you commented.Message ID:
***@***.***>
|
I've been trying to train a model on an early subset of the SPICE dataset. All my efforts so far have been unsuccessful. I must be doing something wrong, but I really don't know what. I'm hoping someone else can spot the problem. My configuration file is given below. Here's the HDF5 file for the dataset.
I've tried training with or without derivatives. I've tried a range of initial learning rates, with or without warmup. I've tried varying model parameters. I've tried restricting it to only molecules with no formal charges. Nothing makes any difference. In all cases, the loss starts out at about 2e7 and never decreases.
The dataset consists of all SPICE calculations that had been completed when I started working on this a couple of weeks ago. I converted the units so positions are in Angstroms and energies in kJ/mol. I also subtracted off per-atom energies. Atom types are the union of element and formal charge. Here's the mapping:
If anyone can provide insight, I'll be very grateful!
The text was updated successfully, but these errors were encountered: