Looping a script #325
Comments
|
Actually I just realised I dont add --mask to the snippy command first I use it after I run snippy! Sorry! |
|
Actually I need to reopen this. How do I tell snippy to add the output of each sample to the same output folder? It wants to replace the first file with the subsequent ones. Is there anyway to tell it in the script above to just keep adding? Cheers |
|
Actually, figured it out. I use ${sample} as the end path in my --outdir command! |
|
@peflanag have you considered using https://github.com/tseemann/snippy/blob/master/README.md#using-snippy-multi Also make sure you are using |
|
Hi @tseemann I haven't cause I had a .sh file with the names of the files in it so for ease of hand I didn't want to rewrite everything in excel to make the tab file. But also, I'm kind of uncertain how I go about it. the manual says the isolates have to be labelled Isolate1 Isolate1b and so on. But I have more samples than the number of the alphabet, over 120 samples. So, do i go from ...Isolate1z to Isolate2 Isolate2b...? But on the manual the Isolate2 seems to correspond to single end reads. I'm basically trying to compare MTBseq and Snippy but cant seem to get either to finish locally or on a college cluster. I'm running snippy 4.4.3 Cheers, P |
|
You do not need to name the isolates that way. |
|
Oh right. So I just write the "name" and then "path" in excel for each sample? I didn't know you can write scripts to generate the input.tab file automatically. Is that something you can share? Or is there somewhere online I can look to explain that? I'm not really tech savvy when it comes to writing scripts. I'm a microbiologist! Cheers, P |
|
For your case, you need a 3 column Excel spreadsheet
Save it as 'tab delimited text' (usually .txt extension) PS. there is no magic script. you could achieve the same by using excel functions probably, as long as the file names are named consistently. |
|
Cheers. Just wondering dos2unix, is that if I am using windows? I'm using a Mac |
|
Oh and do I put the title names ID etc in the tops of the columns or leave them out? |
|
No titles. Just like the docs show. |
|
cheers! |
|
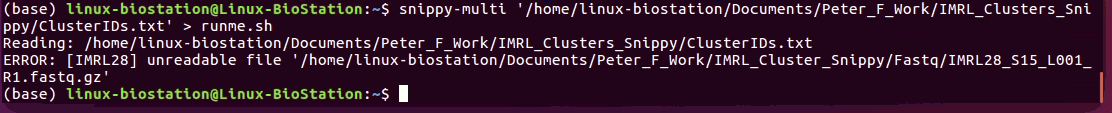
Hey @tseemann sorry to bother you again. I've made the tab file and I ran the script but it said the sample was an unreadable file. Any idea what might be wrong? Cheers, P
|
|
I should point out that this is the first file in the folder so I'm guessing all the others will have the same errors |
|
Hi @tseemann just wondering if you happened to see the message above? All the best, Peter |
|
Snippy can't read that file. Usually this is
|
|
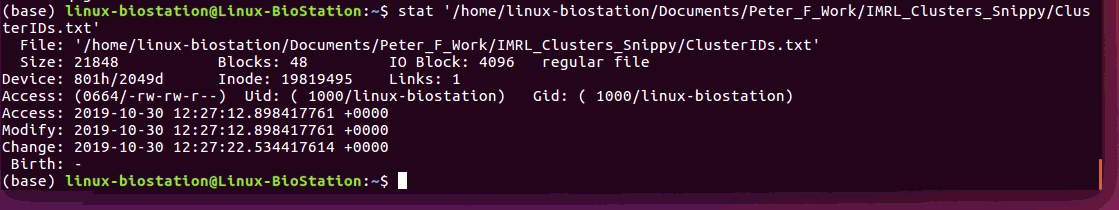
Hi @tseemann I ran the stat /Path/To/File and attached a screenshot below. Snippy is running v4.4.3 I'm not sure what a Windows Linux Subsystem is. I have a macOS machine and a Ubuntu machine. Any of the PCs in work are restricted and I don't have command line access on them. I made the file on excel on my mac, saved as tab delimitated which made a text file and I copied it to the Linux system to run.
|
|
|
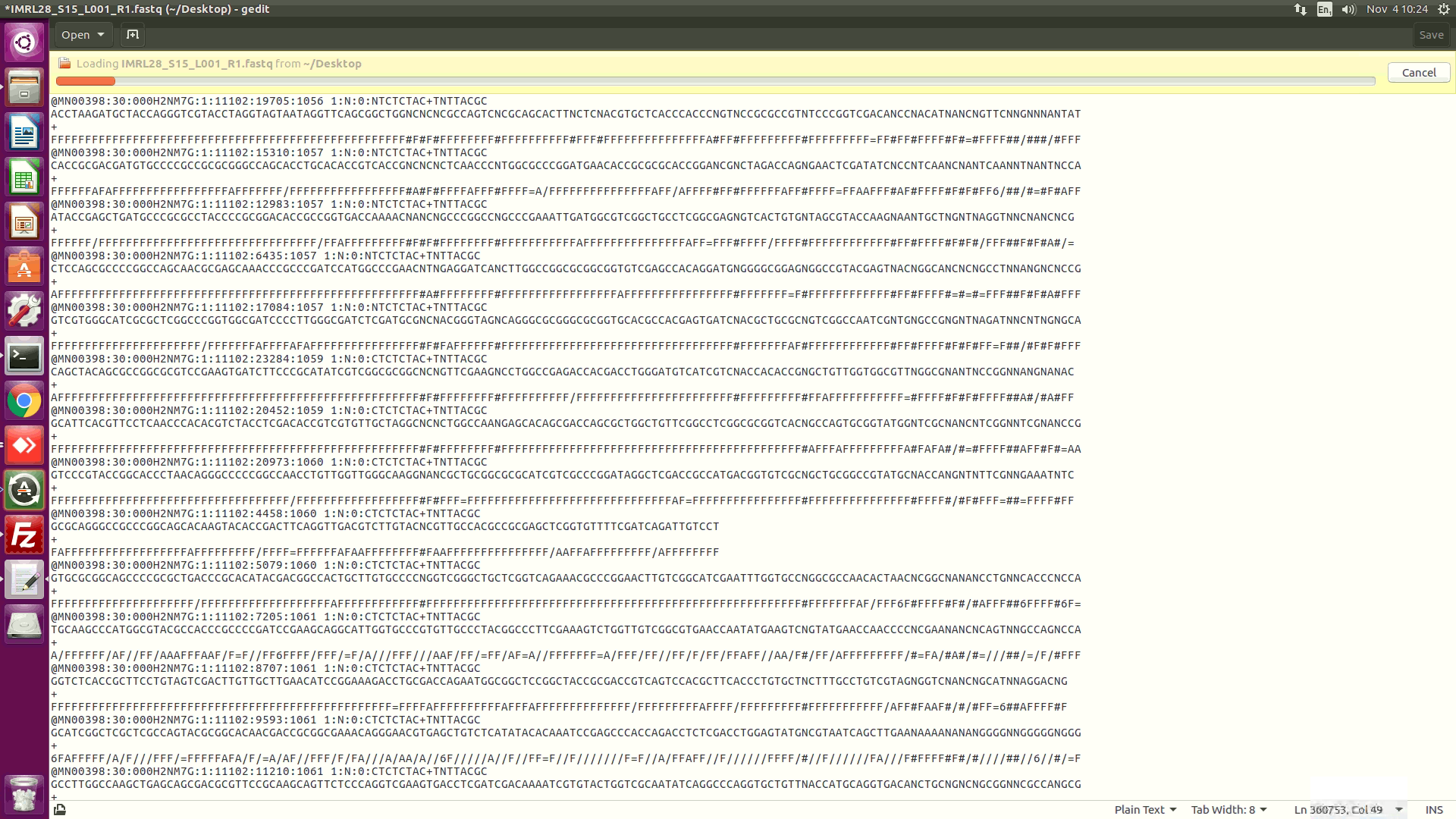
Hi @tseemann I ran mac2unix but it didnt work. I have attached a screenshot of the MiniSeq compressed fastq file that I opened. I'm not sure how it should look for snippy though.
|
|
No, i mean run |
|
I ran “mac2unix tab.file” and it didn’t work. The screenshot is just to show you what the compressed R1 file looks like in case there’s something wrong with that? |
|
It looks fine.
|
|
Hey @tseemann so i did the brew install version of snippy thats on the github page and then when running I ran this command, snippy-multi Path/To/Tab.text --ref Path/To/Ref --mask Path/To/mask.bed > runme.sh and I also tried without the --mask command. I appreciate the help. |
|
Ok, i can see |
|
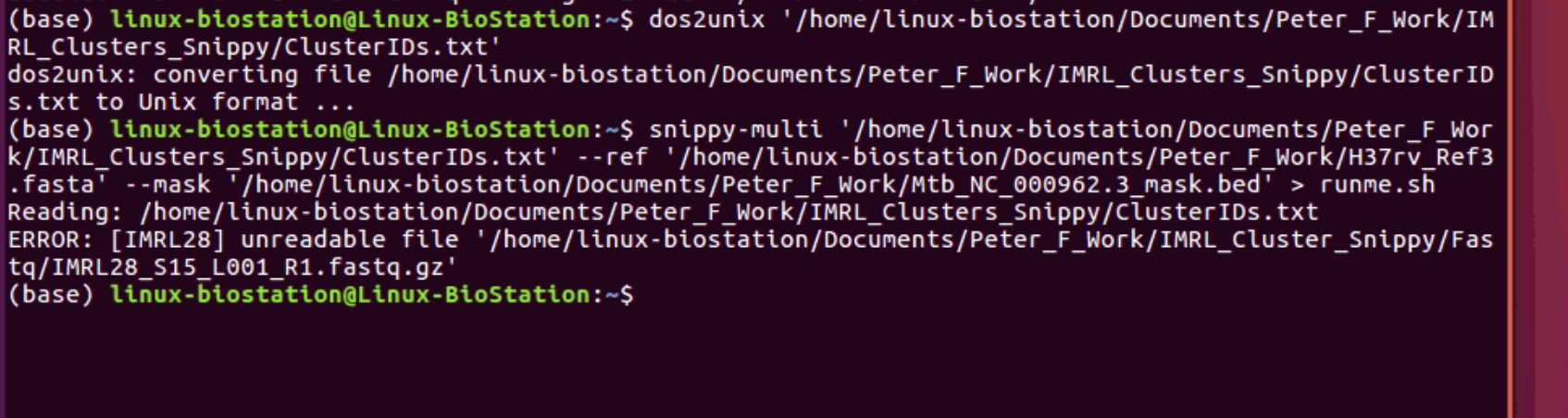
Hey @tseemann so I tried that but again it seems to fail! I don't know what I'm doing wrong? should I try making the tab file on the linux machine instead?
|
|
So it turns out making the file on the Linux machine works but know I have a new error! Any ideas what this means? I don't know what it means by missing read/contig data. Cheers, P
|

|
You seem to have created a CSV file (comma separated) not TSV (tab separated). This hack might fix it: |
|
Hey @tseemann so I tried that but it still hasn't worked. I have attached a screenshot below. I have received a new iMac Pro that is solely for bioinformatics since the linux machine I am using is quite old. Once I set it up I am going to try that tomorrow and hope for the best! But I thought I would attached the screenshot below to see if I am doing it right! P
|

|
You seem to have your old problem back. |
|
Sure, its this: |
|
I don't know why it has a line through it. I am logged into the linux machine from home so trying to paste it from there |
|
When I ran this command, tr "," "\t" < ClusterIDs.csv > ClusterIDs.tab there was no error. it just returned back to (base) linux-biostation@Linux-BioStation:$ |
|
What does this say |
|
Hey @tseemann I ran the the script above and got this: Not sure what it means. I've attached a screenshot cause it was highlighted in red.
|
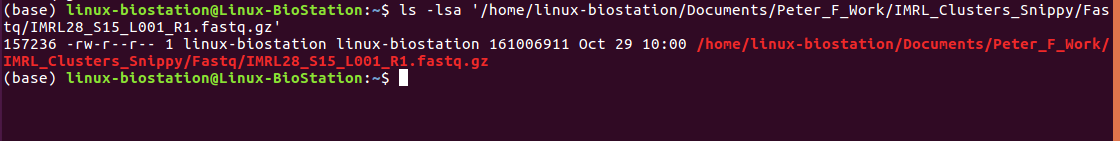
|
The file seems ok. It's red because it's gzip compressed I think. |
|
I’m running it on a Mac this time so I didn’t run mac2unix or dox2unix |
|
You still need to run Details: basically all your text files must have |
|
HI @tseemann cheers for that! I didnt know I still had to do on a Mac. I tried mac2unix "file" and dos2unix" on the file on the mac but got the reply -bash: mac2unix: command not found and the same for dos2unix. Do I have to install something to run it? Cheers |
|
Yes, you need to install those tools somehow (assuming Nullarbor is still not working). I don't know if they are in Conda or not. Sorry. Or copy the file to a Linux system and run it there. |
|
So I managed to install it through Homebrew and ran the snippy command again but I'm still getting the same error. I have pasted it below. Also when I ran od -a file | head -n 20 it looks like the mac2unix or dos2unix didnt work |
|
I also tried it on the linux machine and I don't think it worked. |
|
The file looks fine on the Unix. Maybe I am wrong and you need to leave it Mac format when running on a Mac? Can you run |
|
Hey @tseemann so I ran that IMRL28 sample on the linux environment and it ran fine but when I go to run the snippy-multi it just doesn't like it! I haven't tried on the iMac Pro yet because I just discovered last night when trying to run MTBseq that with the macOS Catalina update, Apple seems to have done something where root permissions are only read and not write and data is on a separate "virtual?" HD. I don't quite understand it but from what I've been reading its messing up conda and conda envs. Needless to say I have wiped the iMac Pro to start everything from scratch. But I dont understand why the file wont work on the Linux machine. I have about 125 samples to run. Just to check, I'm using excel to make the file as attached. Then I click say as and select tab-deliminated which saves as a text file then I'm using mac2unix "file" or dos2unix -c mac "file" |
|
If you are running it on MacOS can you try NOT using |
|
Hey, I’ll try that tomorrow. I’ll save the excel file as a tab-delimimated file which will be .txt and run snippy-multi. If that doesn’t work then I’ll try nullarbor. I was having issues with conda after the macOS Catalina update so hopefully it’ll install. Currently running snippy manually in each sample! |
|
hi @tseemann so i tried what you suggested above and it still doesn't work. This is the print out of my terminal: (nullarbor) peterflanagan@med176028 Clusters_Snippy % nullarbor.pl --ref /Users/peterflanagan/IMRL_Sequencing/Illumina/Clusters_Snippy/H37rv_Ref3.fasta --input /Users/peterflanagan/IMRL_Sequencing/Illumina/Clusters_Snippy/MacClusters.txt --name IMRL_Clusters --outdir /Users/peterflanagan/IMRL_Sequencing/Illumina/Clusters_Snippy/Output I have however managed to write a .sh loop to run snippy and it works. The only problem is it doesnt generate a .vcf file and I dont know why? So I can't run snippy-core afterwards. I have attached teh .sh loop that I wrote. Maybe I left something out? If I run snippy manually on a sample then it makes the vcf file. Peter |
|
Your shell script seems fine. Are you saying NO |
|
Yup! It generates everything in the folders for each sample except a .vcf file |
|
What does the {sample_name}/snippy.log file say? |
|
Hey @tseemann so theres no snippy.log file but there is a snps.log file? I have attached it below and a screenshot of what is in the folder. echo snippy 4.4.5cd /Users/peterflanagan/IMRL_Sequencing/Illumina/Clusters_Snippy/usr/local/bin/snippy --cpus 16 --outdir /Users/peterflanagan/IMRL_Sequencing/Illumina/Clusters_Snippy/Output/IMRL28 --ref /Users/peterflanagan/IMRL_Sequencing/Illumina/Clusters_Snippy/H37rv_Ref3.fasta --R1 /Users/peterflanagan/IMRL_Sequencing/Illumina/Clusters_Snippy/Fastq/IMRL28_S15_L001_R1.fastq.gz -R2 /Users/peterflanagan/IMRL_Sequencing/Illumina/Clusters_Snippy/Fastq/IMRL28_S15_L001_R2.fastq.gzsamtools faidx reference/ref.fabwa index reference/ref.fa[bwa_index] Pack FASTA... 0.03 sec mkdir -p reference/genomes && cp -f reference/ref.fa reference/genomes/ref.faln -sf reference/ref.fa .ln -sf reference/ref.fa.fai .mkdir -p reference/ref && gzip -c reference/ref.gff > reference/ref/genes.gff.gzbwa mem -Y -M -R '@rg\tID:IMRL28\tSM:IMRL28' -t 16 reference/ref.fa /Users/peterflanagan/IMRL_Sequencing/Illumina/Clusters_Snippy/Fastq/IMRL28_S15_L001_R1.fastq.gz /Users/peterflanagan/IMRL_Sequencing/Illumina/Clusters_Snippy/Fastq/IMRL28_S15_L001_R2.fastq.gz | samclip --max 10 --ref reference/ref.fa.fai | samtools sort -n -l 0 -T /private/var/folders/yw/9tmv96yx73ld2r712hclgjv00000gn/T --threads 15 -m 266M | samtools fixmate -m - - | samtools sort -l 0 -T /private/var/folders/yw/9tmv96yx73ld2r712hclgjv00000gn/T --threads 15 -m 266M | samtools markdup -T /private/var/folders/yw/9tmv96yx73ld2r712hclgjv00000gn/T -r -s - - > snps.bamREAD 3922924 WRITTEN 3768726 samtools index snps.bamfasta_generate_regions.py reference/ref.fa.fai 144681 > reference/ref.txtFile "/usr/local/bin/fasta_generate_regions.py", line 7
|
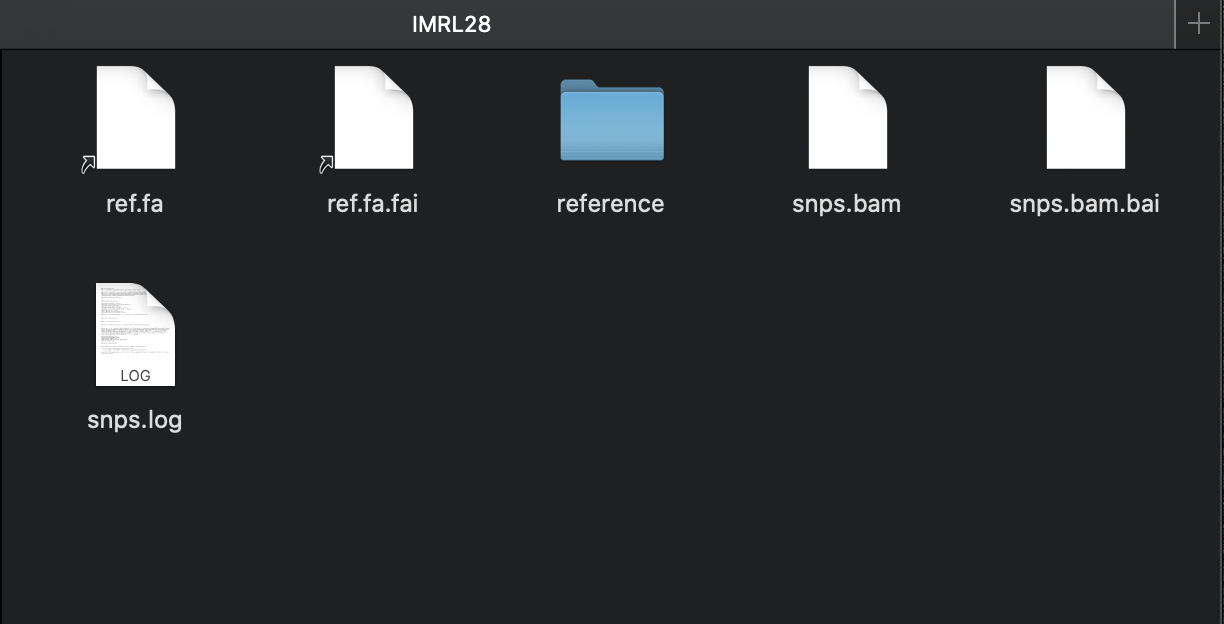
|
You have a problem with python2 vs python3. I think the only way you will get this working is to create a brand new miniconda3 installaiton on linux and install snippy from bioconda. follow these precisely: https://bioconda.github.io/user/install.html#install-conda |
|
Cool I’ll uninstall miniconda 3 tomorrow and reinstall as above and test! |
|
@tseemann you're a legend! I dont want to jinx it but it seems to be working and making all the required files in the folders now! Thanks for your help!
|
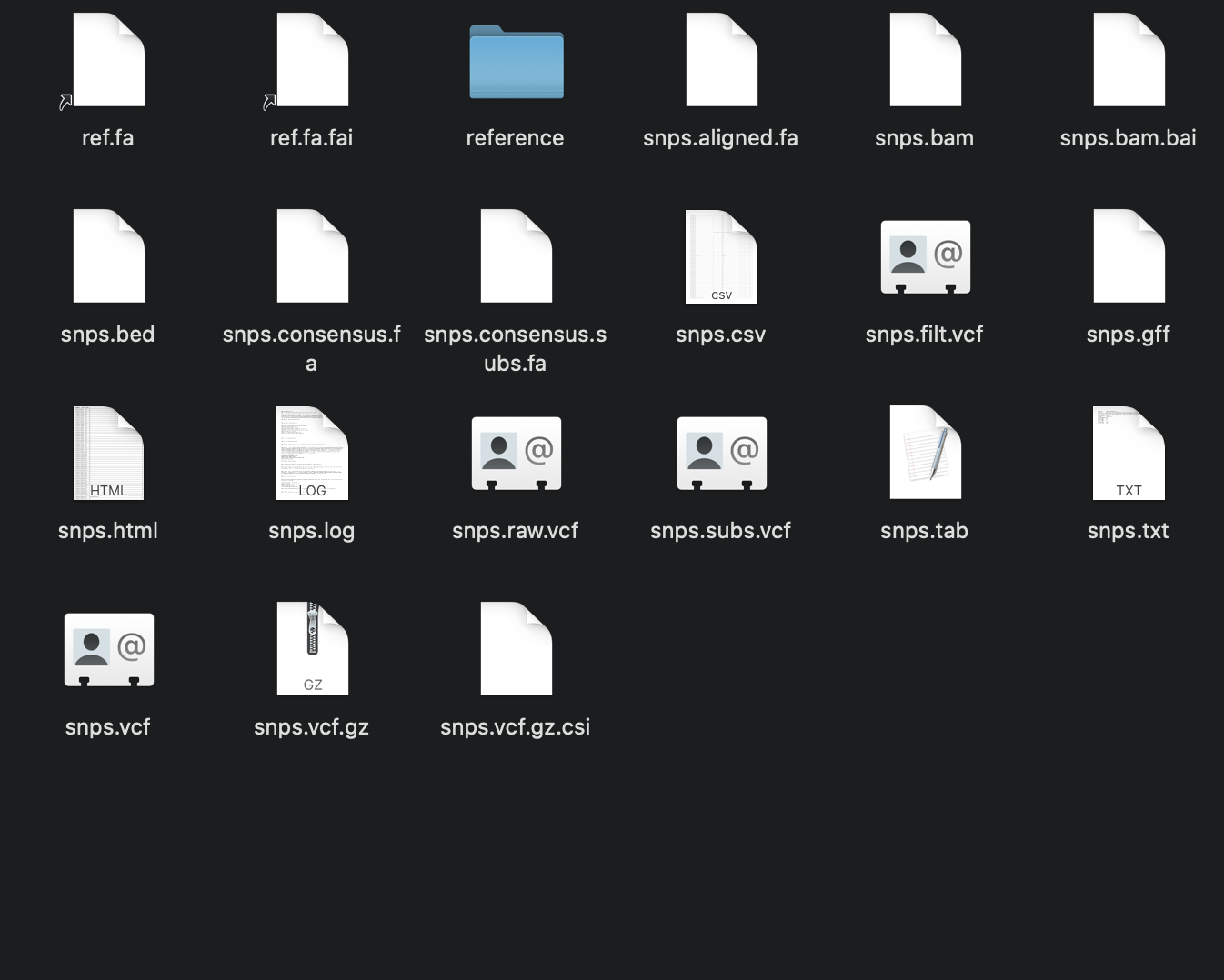
Hi,
I have quite a few file I want to run through Snippy. I made a file that I made executable using chmod u+x and then ran ./MyFile
However it seems to have failed saying "unknown option: mask" I have copied my executable below. It would be great if someone could tell me what I'm doing wrong.
Cheers.
#/bin/bash
sampleLoc=/home/linux-biostation/Documents/Peter_F_Work/IMRL_Clusters_Snippy/Fastq
sampleName=( "IMRL28" "IMRL29" "IMRL30" "IMRL137" "IMRL138" "IMRL39" "IMRL140" "IMRL141" "IMRL142" "MABC143" "IMRL144" "IMRL145" "IMRL146" "IMRL147" "IMRL148" "IMRL149" "IMRL150" "IMRL151" "IMRL152" "IMRL153" "IMRL154" "IMRL156" "IMRL157" "IMRL158" "IMRL159" "IMRL160" "IMRL161" "IMRL162" "IMRL163" "IMRL164" "IMRL165" "IMRL167" "IMRL168" "IMRL169" "IMRL170" "IMRL171" "IMRL172" "IMRL173" "IMRL174" "IMRL175" "IMRL176" "IMRL177" "IMRL178" "IMRL179" "IMRL180" "IMRL181" "IMRL182" "IMRL183" "IMRL184" "IMRL185" "IMRL186" "IMRL187" "IMRL188" "IMRL189" "IMRL190" "IMRL190" "IMRL191" "IMRL192" "IMRL193" "IMRL194" "IMRL194" "IMRL195" "IMRL196" "IMRL197" "IMRL198" "IMRL199" "IMRL200" "IMRL201" "IMRL202" "IMRL203" "IMRL204" "IMRL205" "IMRL206" "IMRL207" "IMRL208" "IMRL209" "IMRL210" "IMRL211" "IMRL212" "IMRL213" "IMRL214" "IMRL215" "IMRL216" "IMRL217" "IMRL219" "IMRL220" "IMRL221" "IMRL222" "IMRL223" "IMRL225" "IMRL226" "IMRL228" "IMRL232" "IMRL233" "IMRL234" "IMRL235" "IMRL236" "IMRL237" "IMRL238" "IMRL239" "IMRL241" "IMRL243" "IMRL244" "IMRL251" "IMRL252" "IMRL253" "IMRL254" )
for sample in ${sampleName[*]}
do
snippy --outdir /home/linux-biostation/Documents/Peter_F_Work/IMRL_Clusters_Snippy/Snippy_Output/ --ref /home/linux-biostation/Documents/Peter_F_Work/H37rv_Ref3.fasta --mask /home/linux-biostation/Documents/Peter_F_Work/Mtb_NC_000962.3_mask.bed --R1$sampleLoc/$ {sample}R1.fastq.gz --R2 $sampleLoc/$ {sample}R2.fastq.gz
done
The text was updated successfully, but these errors were encountered: