Next-generation sequencing has revolutionized the study of cancer genomes. However, the reads obtained from next- generation sequencing of tumor samples often consist of a mixture of normal and tumor cells, which themselves can be of multiple clonal types. A prominent problem in the analysis of cancer genome sequencing data is deconvolving the mixture to identify the reads associated with tumor cells or a particular subclone of tumor cells. Solving the problem is, however, challenging due to the so-called “identifiability problem”, where different combinations of tumor purity and ploidy often explain the sequencing data equally well. Here, we propose a new model to resolve the identifiability problem by integrating two types of sequencing information - somatic copy number alterations and loss of heterozygosity - within an unified probabilistic framework. We derive algorithms to solve our model, and implement them in a software package called PyLOH. We also introduce a novel visualization method "BAF heat map" to to characterize the cluster pattern of LOH. If you have any questions, please email yil8@uci.edu
Mandatory
-
Python (2.7). Python 2.7.3 is recommended.
-
Numpy(>=1.6.1). You can download the source of Numpy from here.
-
Scipy(>=0.10). You can download the source of Scipy from here.
-
Pysam(>=0.7). Pysam_0.7X preferred, Pysam_0.8X tested and seems to be much slower (preprocessing the example data with 5 cores Intel i7, Pysam_0.7.5 took ~4 hours and Pysam_0.8.4 took ~17 hours). To install Pysam, you also need to install Cython first.
Optional
- matplotlib(>=1.2.0) is required to plot BAF heat map.
Although not mandatory, Linux system is recommended. Also, samtools is not required by PyLOH, but can be useful for creating bam, bam index and fasta index files which are required by the pysam module of PyLOH.
Download the compressed source file PyLOH-*.tar.gz and do as follows:
$ tar -xzvf PyLOH-*.tar.gz
$ cd PyLOH-*
$ python setup.py install
If you prefer to install PyLOH other than the default directory, you can also use this command:
$ python setup.py install --prefix /home/yili/
There are also config/ and bin/ folders under PyLOH-*. The config/ folder contains example priors and the bin/ folder contains
useful utilities, such as the R code to run BICseq, DNAcopy and the python script to
convert BICseq/DNAcopy results to BED file. You can copy these two folders somewhere easily accessible.
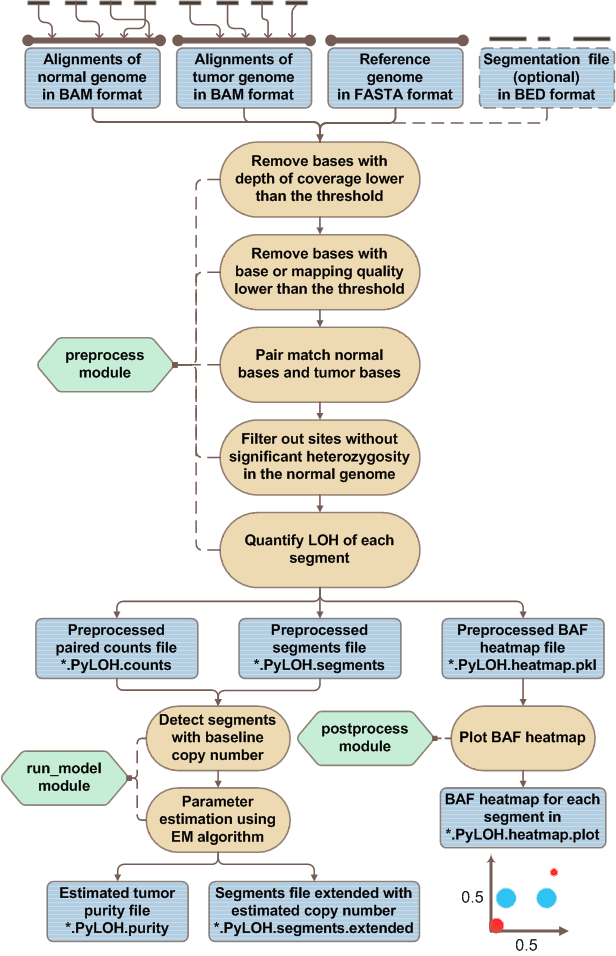
PyLOH is composed of three modules:
-
preprocess. Preprocess the reads aliments of paired normal-tumor samples in BAM format and produce the paired counts file, preprocessed segments file and preprocessed BAF heat map file as output. -
run_model. Take the paired counts file and preprocessed segments file as input, estimate tumor purity, the copy number of each segment. -
postprocess. Take the preprocessed BAF heat map file as input and plot the BAF heat map for each segment as output.
The general workflow of PyLOH is this
This part of README is based on JoinSNVMix. To preprocess the paired cancer sequencing data, execute:
$ PyLOH.py preprocess REFERENCE_GENOME.fasta NORMAL.bam TUMOUR.bam BASENAME --segments_bed SEGMENTS.bed --min_depth 20 --min_base_qual 10 --min_map_qual 10 --process_num 10
REFERENCE_GENOME.fasta The path to the fasta file that the paired BAM files aligned to. Currently, only the UCSC and ENSEMBL chromosome format are supported. Note that the index file should be generated for the reference genome. This can be done by running samtools as follows:
$ samtools faidx REFERENCE_GENOME.fasta
NORMAL.bam The BAM file for the normal sample. The BAM index file should be generated for this file and named NORMAL.bam.bai. This can be done by running
$ samtools index NORMAL.bam
TUMOUR.bam The bam file for the tumour sample. As for the normal this file needs to be indexed.
BASENAME The base name of preprocessed files to be created.
--segments_bed Use the genome segmentation stored in SEGMENTS.bed. If not provided, use 22 autosomes as the segmentaion. But using automatic segmentation algorithm to generate SEGMENTS.bed is highly recommended, such as BICseq for whole genome sequencing data and DNAcopy for whole exome sequencing data.
--WES Flag indicating whether the BAM files are whole exome sequencing(WES) or not. If not provided, the BAM files are assumed to be whole genome sequencing(WGS).
--min_depth Minimum depth in both normal and tumor sample required to use a site in the analysis.
--min_base_qual Minimum base quality required for each base.
--min_map_qual Minimum mapping quality required for each base.
--process_num Number of processes to launch for preprocessing.
After the paired cancer sequencing data is preprocessed, we can run the probabilistic model of PyLOH by execute:
$ PyLOH.py run_model BASENAME --allelenumber_max 2 --max_iters 100 --stop_value 1e-7
BASENAME The base name of preprocessed files created in the preprocess step.
--allelenumber_max The maximum copy number of each allele allows to take.
--priors Path to the file of the prior distribution. The prior file must be consistent with the --allele_number_max. If not provided, use uniform prior, which is recommended.
--max_iters Maximum number of iterations for training.
--stop_value Stop value of the EM algorithm for training. If the change of log-likelihood is lower than this value, stop training.
Currently, the postprocess module is only for plotting the BAF heat map of each segment:
$ PyLOH.py BAF_heatmap BASENAME
BASENAME The base name of preprocessed files created in the preprocess step.
*.PyLOH.counts The preprocessed paired counts file. It contains the allelic counts information of sites, which are heterozygous loci in the normal genome. The definition of each column in a *.PyLOH.counts file is listed here:
| Column | Definition |
|---|---|
| seg_index | Index of each segment |
| normal_A | Count of bases match A allele in the normal sample |
| normal_B | Count of bases match B allele in the normal sample |
| tumor_A | Count of bases match A allele in the tumor sample |
| tumor_B | Count of bases match B allele in the tumor sample |
*.PyLOH.segments The preprocessed segments file. It contains the genomic information of each segment. The definition of each column in a *.PyLOH.segments file is listed here:
| Column | Definition |
|---|---|
| seg_name | Name of the segment |
| chrom | Chromosome of the segment |
| start | Start position of the segment |
| end | End position of the segment |
| normal_reads_num | Count of reads mapped to the segment in the normal sample |
| tumor_reads_num | Count of reads mapped to the segment in the tumor sample |
| LOH_frec | Fraction of LOH sites in the segment |
| LOH_status | FALSE -> no LOH; TRUE -> significant LOH; UNCERTAIN -> medium level LOH |
| log2_ratio | Log2 ratio between tumor_reads_num and normal_reads_num |
*.PyLOH.segments.extended The extended segments file after run_model. There are one additional column:
| Column | Definition |
|---|---|
| copy_number | Estimated copy number of the segment |
*.PyLOH.purity Estimated tumor purity.
*.PyLOH.heatmap.pkl The preprocessed BAF heat map file in Python pickle format.
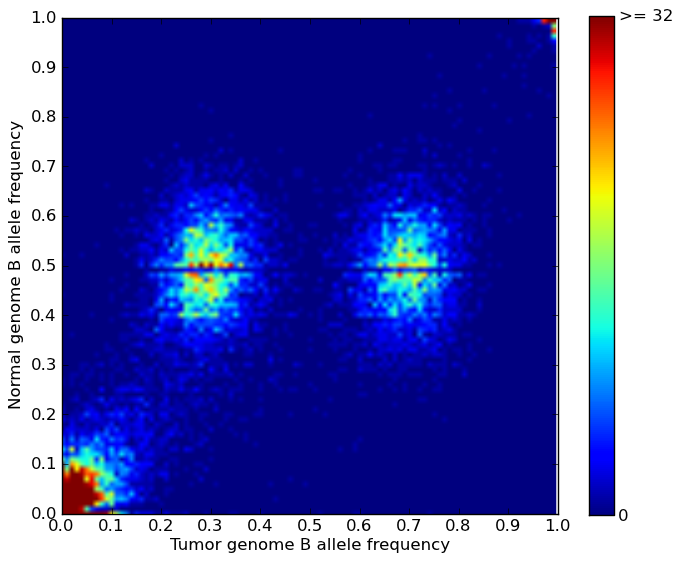
*.PyLOH.heatmap.plot The folder of BAF heat maps plotted for each segment. A typical BAF heat map looks like this
We highly recommend using automatic segmentation algorithm to partition the tumor genome, and thus prepare the segments file in BED format.
For exmaple, we used BICseq in the original paper for whole genome sequencing (WGS) data. To run a BICseq analysis, you
can copy the commands in bin/BICseq.R and paste them in a R interative shell. Or you can also run the R script from the command line:
$ R CMD BATCH bin/BICseq.R
Note that,normal.bam and tumor.bam must be in the same directory where you run the command. The R script will output a segments file
segments.BICseq. Then you can use the script bin/seg2bed.py to convert the segments file into BED format:
$ seg2bed.py segments.BICseq segments.bed --seg_length 1000000
--seg_length Only convert segments with length longer than the threshold.
For whole exome sequencing (WES) data, since reads coverage on targeted exonic regions are no longer randomly
distributed due to probe's variable effciency, DNAcopy is recommended for segmentation instead of BICseq. To run DNAcopy analysis, you can firstly use the script bin/bam2DNAcopy.py to convert the paired BAM files of the normal and tumor sample into the input file for DNAcopy:
$ bam2DNAcopy.py NORMAL.bam TUMOUR.bam EXONS.bed DNAcopy.bed --min_depth 100
EXONS.bed The input bed file for all exon regions. Examples from Illumina TruSeq are included under data/.
DNAcopy.bed The output bed file for running DNAcopy.
--min_depth Minimum reads detph required for each exon region in both normal and tumor samples. Default is 100.
Then you can run bin/DNAcopy.R the same way as bin/BICseq.R. Again, DNAcopy.bed must be in the same directory
where you run the command. The R script will output a segments file segments.DNAcopy. Finally you can also use the script bin/seg2bed.py to convert the segments file into BED format then same way as for BICseq.
We used Homo_sapiens_assembly18.fasta from Broad Institute as the REFERENCE_GENOME.fasta in the original paper. One example of simulation data can be found here. Please use binary mode to download the BAM file(e.g. wget command), directly ftp download using browser may corrupt the BAM file. The ground truth tumor purity of this data is 90%.
Li, Y., Xie, X. (2014). Deconvolving tumor purity and ploidy by integrating copy number alterations and loss of heterozygosity. Bioinformatics.