1.5.14 Release Notes
- Layout and Depiction
- Simpler API
- Alignment
- Rxn Auto Align
- Molecular Formula
- Round Robin Algorithm
- SMILES
- SmiFlavor
- CXSMILES
- Pattern Matching
- Reaction queries and matching
- SMARTS Fragment Extraction
IAtomContainer mol = ...;
IAtomContainer rxn = ...;
StructureDiagramGenerator sdg = new StructureDiagramGenerator();
sdg.generateCoordinates(mol); // mol layout
sdg.generateCoordinates(rxn); // rxn layoutPreviously...
IAtomContainer mol = ...;
StructureDiagramGenerator sdg = new StructureDiagramGenerator();
sdg.setMolecule(mol); // implicit clone of molecule
sdg.generateCoordinates();
mol = sdg.getMolecule();
for (IAtomContainer comp : ReactionManipulator.getAllAtomContainers(rxn)){
sdg.setMolecule(comp, false); // if the molecule is clone it's a pain to set it back to the reaction!
sdg.generateCoordinates();
}The SDG can can be told to keep certain atoms or bonds fixed in position. The fixing of atoms controls whether their coordinates changes, whilst fixing bonds disables the stretching to avoid overlaps etc. The API is low level but powerful.
Basic usage:
IAtomContainer mol = ...;
Set<IAtom> afix = new HashSet<>();
Set<IBond> bfix = new HashSet<>();
mol.getAtom(0).setPoint2d(new Point2d(0, 1));
mol.getAtom(1).setPoint2d(new Point2d(0, 2));
afix.add(mol.getAtom(0));
afix.add(mol.getAtom(1));
StructureDiagramGenerator sdg = new StructureDiagramGenerator();
sdg.setMolecule(mol, false, afix, bfix); // note clone disabled!
sdg.generateCoordinates();Example aligning MCS search results, it's a little bit hard to see as the ring aligns to a template on the left and looks good already, but notice on the right how the halogens align.
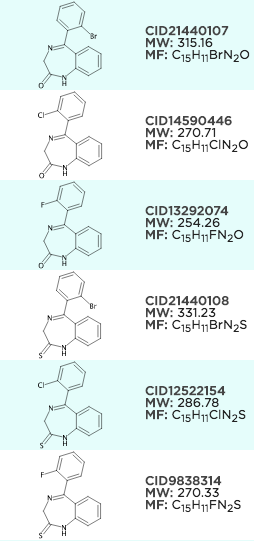
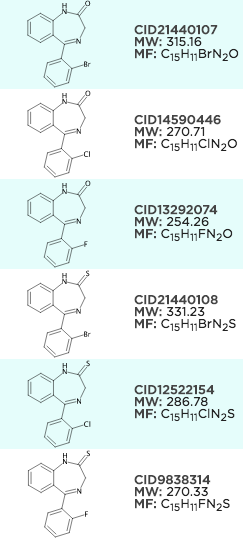
Since we're retro fitting some old code to do this, there may be a few quirks but these can normally be rectified with an provided example.
When a reaction has an atom mapping, it's possible to automatically align the coordinates on the left and right reasonably cheaply. This option has therefore be added to the structure diagram generator.
Default:
Aligned:
To enable/disable alignment use:
StructureDiagramGenerator sdg;
sdg.setAlignMappedReaction(false);Included semi-empirical generation that extends a provided dictionary.
Neither SnCl2 or HOOH is in the provided dictionary:
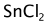
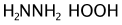
Boc is in dictionary but is collapsed back to NHBoc:
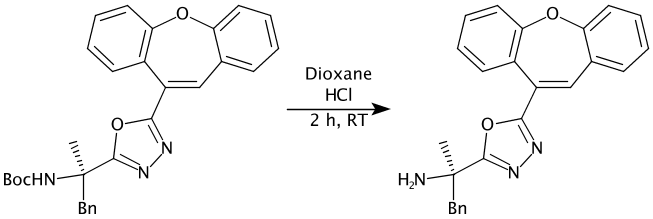
See pull request 210 for more details.
Isotopes ifac = Isotopes.getInstance();
MolecularFormulaRange range = new MolecularFormulaRange();
range.addIsotope(ifac.getMajorIsotope("C"), 8, 20);
range.addIsotope(ifac.getMajorIsotope("H"), 0, 20);
range.addIsotope(ifac.getMajorIsotope("O"), 0, 1);
range.addIsotope(ifac.getMajorIsotope("N"), 0, 1);
MolecularFormulaGenerator tool = new MolecularFormulaGenerator(SilentChemObjectBuilder.getInstance(),
133.0, 133.1, range);
IMolecularFormulaSet mfSet = tool.getAllFormulas();
for (IMolecularFormula mf : mfSet.molecularFormulas()) {
System.out.println(MolecularFormulaManipulator.getString(mf) + " " +
MolecularFormulaManipulator.getTotalExactMass(mf));
}The recent SMILES API has previously used the terms generic, unique, isomeric, absolute to control the flavor of SMILES generated. The meanings of theses terms aren't always obvious, for example, 'unique' does not include stereochemistry, and different between toolkits, 'absolute' and 'isomeric' and have their meanings swapped in OEChem compared to Daylight (which CDK matched here).
SmilesGenerator smigen = SmilesGenerator.unique();This will still work but the new way of configuring SMILES is with binary flags specified in SmiFlavor. These flags give
fine control over what is written in the output SMILES, for example I can enable stereo chemistry to varying levels:
SmilesGenerator smigen = new SmilesGenerator(SmiFlavor.Stereo); // all stereo on
SmilesGenerator smigen = new SmilesGenerator(SmiFlavor.StereoTetrahedral); // only tetrahedral stereo on
// turn on Stereochemistry and AtomicMass, non-canonical
SmilesGenerator smigen = new SmilesGenerator(SmiFlavor.Stereo | SmiFlavor.AtomicMass);ChemAxon Extended SMILES can now also be written, one of the many extensions it provides is to store coordinates. As above, the SmiFlavor controls which parts of the CXSMILES is written. CXSMILES is written by default - normall
String smi = "CC* |$;;R1$,(0,0,;1,0,;2,0,;)|"
SmilesParser smipar = new SmilesParser(SilentChemObjectBuilder.getInstance());
IAtomContainer mol = smipar.parse(smi);
new SmilesGenerator(SmiFlavor.Default).create(mol);
// CC* |$;;R1$|
new SmilesGenerator(SmiFlavor.CxSmilesWithCoords).create(mol);
// CC* |$;;R1$,(0,0,;1,0,;2,0,;)|The Pattern API has been extended allowing to match reactions and accept reaction SMARTS. This includes matching
the semantics of the atom-atom mapping matching.
Pattern ptrn = SmartsPattern.create("[N:1]C(=O)OC(C)(C)C>>[NH:1]"); // a Boc deprotection
String rsmi = "CC(C)(C)OC(=O)[N:5]1[CH2:4][CH2:3][N:2]([CH2:7][CH2:6]1)[CH2:1][C:8]#[CH:9]>C(Cl)Cl.C(=O)(C(F)(F)F)O>[CH:9]#[C:8][CH2:1][N:2]1[CH2:3][CH2:4][NH:5][CH2:6][CH2:7]1";
SmilesParser smipar = new SmilesParser(SilentChemObjectBuilder.getInstance());
IReaction rxn = smipar.parseReactionSmiles(rsmi);
if (ptrn.matches(rxn))
{
// hit
}
Matching of not reaction queries also works.
Pattern ptrn = SmartsPattern.create("*!-*"); // non-sigma bond
String rsmi = "CC(C)(C)OC(=O)[N:5]1[CH2:4][CH2:3][N:2]([CH2:7][CH2:6]1)[CH2:1][C:8]#[CH:9]>C(Cl)Cl.C(=O)(C(F)(F)F)O>[CH:9]#[C:8][CH2:1][N:2]1[CH2:3][CH2:4][NH:5][CH2:6][CH2:7]1";
SmilesParser smipar = new SmilesParser(SilentChemObjectBuilder.getInstance());
IReaction rxn = smipar.parseReactionSmiles(rsmi);
if (ptrn.matches(rxn))
{
// hit
}
A SmartsFragmentExtractor has been added that allow the generation of SMARTS that matches a provided set of atoms (atom index):
IChemObjectBuilder bldr = SilentChemObjectBuilder.getInstance();
SmilesParser smipar = new SmilesParser(bldr);
IAtomContainer mol = smipar.parseSmiles("[nH]1ccc2c1cccc2");
SmartsFragmentExtractor subsmarts = new SmartsFragmentExtractor(mol);
String smarts = mol.generate(new int[]{0,1,3,4});
// smarts=[nH1v3X3+0][cH1v4X3+0][cH1v4X3+0][cH0v4X3+0] 152 John Mayfield
56 Egon Willighagen 0000-0001-7542-0286
13 Nikolay Kochev
5 Kai Dührkop
3 Tomáš Pluskal
2 Nina Jeliazkova
1 Hsiao Yi
1 Rajarshi Guha- Bumping version for release.
ddfbdb6 - Update .travis.yml
4b478a2 - Merge pull request #245 from cdk/release-prep
dce858e - Merge pull request #244 from egonw/fix/restorePrevBehaviour
40b8dad - Merge pull request #242 from egonw/patch/ignoreLimitations
6e2ca0a - More slow tests.
9449cb7 - Better way to ignore slow tests by default.
07d8b53 - Slow tests are slow - disable by default they hinder build process.
b0da84a - Cache dictionary databases - faster initialisation.
a6b97cd - Ignore non-deterministic test.
f8c8d4c - Regression due to atom type assignment previously removing arom flags.
f3a4c13 - Fix XML validation, current code doesn't make sense since the project is Java 1.7+.
f63e6a1 - Restore the API behaviour: throw an exception for null input
26806f2 - Ionic bonds are now 1.5x length.
e8194ab - AtomType manipulator used to remove aromatic flags, they are now kept and so the fingerprint value has changed.
9ef289d - Don't run these tests, as they are expected to fail
0f0203b - Correct test output, 1 unpaired electron = RAD 2 (doublet)
1f1e7e2 - Merge pull request #231 from cdk/patch/smarts-ecfp
02872f8 - Fix reviewer comments. + license header + citation + resync bib with upstream
6889bf8 - New contributor
fa6f843 - fix some typo in javadoc
4972d07 - Merge pull request #241 from cdk/patch/abbr_rendering
a79da5d - HOOH not OHOH.
2b83fa4 - Forgot other test class - spotted quirk with writing charges which is now resolved.
c575ef1 - Merge pull request #234 from egonw/fix/typos
aee807e - Finalise patch: rename to make it clear what it is doing and is easy to find (start with 'Smarts' prefix). Introduce the MODE_EXACT to specify setting the exact valence. We use int constants rather than enums to avoid adding two classes to the public API.
144f4d7 - Change functionality - don't write colon (:) for peripheral aromatic bonds, it is redundant.
adff265 - Extract and generalise SMARTS substructure functionality to a separate class. Copies exact functionality ATM.
a43a478 - More verbose unit testing with hamcrest matchers.
93922bf - Minor tidy up - no functionally modified.
efef10c - statis smilesparser for the test
2ae1873 - refactored to junit4
1f4b09e - Added test case for CircularFingerprint smarts
72c4a65 - code cleanup
de46bbf - Implemented handling of aromatic bonds
1bcd340 - Implemented handling of ring closures
080f776 - Fixed a bug in external atom check
2a8e5b1 - Implemented handling of 'external' neighbour atoms
58f9899 - Implemented basic functionality in nodeToString()
6f12927 - set up for recursive traversing of the fingerprint structure
6515a38 - improved the atom smarts
f2e3f55 - improved the atom smarts
d354570 - implemented functions getChargeSmartsStr() and getAtomSmarts()
d6a0fc5 - added function findArrayValue()
f0864b3 - Added new function getFPSmarts()
7a95b98 - Special case should only kick in when the atom count is 2.
953e611 - Add Trt token to abbreviation dictionary.
02b1fa8 - Merge pull request #240 from cdk/patch/inchi_taut_options
4cd5ab5 - Allow contractions like SnCl2.
aaae48e - Tweaks to disconnected component placement to try and avoid overlaps.
91a92e4 - Improved composite abbreviations for mixtures, use interpunct dot rather than slash, possible option in future.
7041beb - Need space after option.
ce1938f - InChI KET and 15T options.
825225b - Merge pull request #239 from cdk/patch/order-reactants
4ac5e5e - Change the order of components when aligned such that it matches the order they are bonded in the product.
0d267c3 - Merge pull request #238 from cdk/patch/abbr-tokens
a9c32b1 - Organise and improve abbreviation tokens.
f2f02e4 - Merge pull request #237 from cdk/patch/improved-abbr-labels
0ce6a8a - Merge pull request #236 from cdk/patch/rxn-comp-grouping
da92110 - Reverse abbreviation labels with numbered brackets correctly, B(OH)2 becomes (HO)2B not 2(HO)B. Add recent reduce method needed for tests to pass.
a3ddccb - Improved abbreviations through collapsing those next to hetroatoms, NH-Boc is collapsed to NHBoc.
a9062d7 - Abbreviation labels can now go above/below the labels.
68d76b3 - Visible neighbours may be more than 1 now.
3c0f5c5 - Allow abbreviations with more than one connection providing it is to the same atom.
3963158 - Don't optimize runs of similarly styled charcters.
fe5a0b5 - Another typo fix
1a7b017 - Fixed spelling error
90df1d8 - Unit test to make sure parsing a SMILES does not set a null cdk:Title (as 1.5.13 does, as found in Bioclipse)
9133fa6 - Ensure component grouping is correctly initialised for reaction queries.
7518992 - Fixed a number of spelling errors
c35b216 - Merge pull request #235 from cdk/hotfix/single-bond-between-arom
35d5741 - Minor quirk of beam internal, a recent update in CDK unsets the bond order when reading non-kekulé SMILES, during the conversion we need to tell the converter this to avoid nulling out single bonds between aromatic atoms.
0d2d59f - Merge pull request #233 from cdk/patch/smiflags-spelling
bb12f67 - Resolve test with modified output - one of these (assignDbStereo) came from the beam algorithm change for assign up/down cis-trans indication.
6feb445 - Default SMILES output.
9cece8b - Use american spelling of flavour.
cae0258 - The Molecule class no longer exists
1906a87 - Small typo fix
8d4d5f2 - Merge pull request #232 from egonw/patch/isotopeJavaDoc
39ffc88 - Extended the JavaDoc to point to the data source (closes #1381)
2845a56 - Merge pull request #221 from cdk/patch/cxsmilesgen
cf622a4 - Merge pull request #230 from cdk/patch/convertH
b5c9719 - Merge pull request #228 from cdk/patch/smarts-stereo-ringclosures
cf60ea5 - Merge pull request #227 from cdk/patch/atomvalueannotations
6f9663f - Merge pull request #223 from cdk/patch/inchitimeout
bfb3d6c - Merge pull request #229 from cdk/patch/beamupgrade
4cdf4eb - Converting implicit to explicit hydrogens needs to copy the stereochemistry [bug:1383]
c05d665 - Allow reading of SMILES extension supported by Beam, atom-based double bond configuration.
c5b5efd - Format elements.
8debe75 - Upgrade Beam to 0.9.2, minor changes and bug fix for Nina's ChEMBL SMILES generation problem.
3c72204 - A SMILES should match it's self as a SMARTS, this example did not. The reason was the stereochemistry ordering was read incorrectly due to how rings were handled in the SMARTS parser, to fix the nesting of the syntax tree needed to be changed.
d9a7de5 - Update SMARTS pattern API - for common usage we normally just pass in null.
67ffd0a - Merge pull request #222 from cdk/patch/mdlrad
6a170cd - Back change that got fixed twice.
a607a73 - Need to use inverse ordering for coordinates.
209208d - Correct way of masking out an option.
69968a8 - Atom values were omitted from output.
c494b38 - New SMILES flavour API documentation.
2dffeff - ChemAxon emits trailing semi-colons for atom labels, this were not correctly parsed.
49939f0 - Output fragment group and correct CXSMILES for reactions, also when canonical.
9f5fb03 - CXSMILES generation for reactions, canonical ordering works correctly.
94f20ab - Unified reaction SMILES API with the molecule and make it so reactions can be canonicalised.
fc5e945 - Consider agents in ReactionManipulator.
c2b88b4 - CXSMILES generation for molecules.
90f6793 - Renamed zCoords field - we will reuse in the writing but not just to indicate z coordinates.
ffc6d52 - Push SmiOpt down to the conversion to beam, this makes things a lot simpler and configurable.
84719a6 - Gearing up for finer control of SMILES output we want to pass in a bitset (composed of SmiOpt flags) into the constructor.
02f3294 - Option to display atom values in depiction generator.
623b350 - Merge pull request #226 from tomas-pluskal/patch-3
6582393 - Added a contributor
2533513 - Merge pull request #225 from tomas-pluskal/patch-2
fadac70 - Update AUTHORS
639ab40 - Better radical and valence support in V3000.
04966f8 - Workaround for timeouts in JNI InChI - default timeout included of 5 seconds rather than infinite.
78af7c0 - Correct MDL radical to/from electron count semantics.
700f2c2 - Merge pull request #220 from egonw/patch/aromatictyOverwriting
2f50ba9 - Merge pull request #219 from egonw/patch/unitTestMultipleSingleElectrons
1097093 - Merge pull request #218 from egonw/patch/missingRef
375d2ec - Merge pull request #217 from egonw/patch/removeCDKSetTags
e1acd01 - Two typo fixes and a few JavaDoc links
1938e0b - Don't configure aromaticity if not explicitly set to true for the atom type (closes #1322)
0dda78a - Test that if the atom type is not explicitly aromatic, it does not overwrite aromaticity information of the configured atom (see bug #1322)
d4b2490 - Also double check the single electron counts (fixes bug #1382)
1f8cd45 - Fixed the string: cannot be the same as another constant (duh)
b06df5a - Created a unit test for bug #1382
c54a210 - Implemented a toString() method for easier debugging
d041f1b - Added reference (closes feature request #115)
92e0d87 - Removed @cdk.set JavaDocs tags for reaction-types (addresses proj maintenance task #70, https://sourceforge.net/p/cdk/project-maintainance-tasks/70/)
5fd1fe3 - Removed @cdk.set JavaDocs tags (addresses proj maintenance task #70, https://sourceforge.net/p/cdk/project-maintainance-tasks/70/)
6c73c37 - Removed @cdk.set JavaDocs tags (addresses proj maintenance task #70, https://sourceforge.net/p/cdk/project-maintainance-tasks/70/)
f62eb0c - Removed @cdk.set JavaDocs tags (addresses proj maintenance task #70, https://sourceforge.net/p/cdk/project-maintainance-tasks/70/)
2765695 - Merge pull request #216 from cdk/patch/pseudoatomhcount
35f580d - Merge pull request #215 from cdk/patch/cxsmiles_atomvals
cc57e73 - Hydrogens should not be suppress on pseudo atoms.
265c98e - Don't reset atom labels when we read atom values and allow trailing ';' in the atom label field.
fc3e65a - Merge pull request #211 from cdk/patch/assign-wedge-bonds
6f16bc5 - Merge pull request #209 from cdk/patch/cxsmi-labelescape
2e9d0de - Merge pull request #213 from cdk/patch/sdgwigglybonds
f33b396 - Merge pull request #214 from tomas-pluskal/molecularformularange
9a777a9 - Added a null check to MolecularFormulaRange
d82fd44 - Can't make this assumption to skip non-planar bond assignment (bold/hash wedges) as we also use it to assign wiggly bonds.
171cb27 - Merge pull request #212 from rajarshi/master
5332a43 - Updated misleading Javadoc
8e6d99f - Merge pull request #210 from kaibioinfo/round_robin
868adff - fix typo
b8a0f44 - Merge remote-tracking branch 'origin/round_robin' into round_robin
17b77da - remove decomp package. Remove all unnecessary classes and methods. Put everything into one class with local access modifier
4f83894 - Improved wedge assignment priority to avoid Sp3 carbons.
8157a18 - remove some remainder of refactoring
6aeeacc - implement round robin algorithm for decomposing molecular formulas taken from SIRIUS
86b4065 - Minor tweak to correctly handle labels where the first character is escaped.
26ed0ab - Merge pull request #208 from cdk/patch/api-fixup
397a0c5 - Modifications to required invariants for tests.
2f2ccc4 - Also need to visit other end of positional variation when moving.
1600782 - When moving around positional variation ensure all atoms are moved.
555cf60 - Use the unset bond order when reading SMILES as non kekulized.
7c77138 - Deprecate default constructor, hinting the caller needs to choose which SMILES to write.
bad6166 - Illegal argument when the new atom already exists in the molecule.
706c84b - Merge pull request #207 from cdk/patch/cxsmi-labelescape
2ade769 - Correct handling of escaped labels.
241f4fe - Added an extension (from a Mopac manual), fixing a failing unit test
38b2f34 - Builder methods in this class are not unit tests
51d39b7 - Newer JUnit version
480e9b6 - Reports were still showing problems in the SMSD code: syntax now according to this answer: http://stackoverflow.com/questions/27225598/how-to-exclude-a-file-for-checking-in-pmd-maven-pom-xml-file
fd324c7 - Ignore two tests, because they cause a circular dependency cycle
08a207b - Updated to Maven PMD 3.6
9a6f578 - Merge pull request #205 from cdk/patch/sdgsublayout
bc1c053 - Remove stray debug statement.
6d9768f - Only include acyclic fixed bonds where the src atoms is also acyclic - adjust refinement to count bonds instead of atoms and allow these to moved out of the way.
0d7a31a - Modification to a previous patch: cis/trans correction should be done before refined by wedges should be done after.
86af265 - Better ringset prioritisation favouring heterocycles
9401728 - Correct handling of partially placed rings.
8675e1b - Simplified (and corrected for partially laid out rings) bridged ring placed, rather than changing the draw direction we just ensure the winding of the bridged atoms and the other share atoms is consistent.
2d98f18 - Tidy up and core method in 2D atom placing - minor optimisation 2N -> N.
8addbe5 - Improve reaction alignment by only transferring coordinates for the largest part.
f9d9c5c - When refining the layout, don't flip parts with fixed atoms.
c1c4ead - Provide new lower-level connected components that allows us to pass in a custom partition.
4241fcf - Layout disconnect fragments closer together.
ca3c29b - Use the labelCoords to centre fragment abbreviations.
d4464be - Improve reaction align, detect when a mapping is likely to produce problems (stoichiometry).
c7df5df - Correct terminal ring peeling.
225c8e4 - Handle abbreviation mixtures.
76bc2a7 - Copy align map rxn param.
858d5c0 - Option to enable/disable mapped reaction alignment.
50b0a7d - Correct layout of partially fixed ring systems.
e16b3da - Pass through afix/bfix when laying out sub fragments/salts.
1e430a1 - Clean up reaction layout in DepictionGenerator, no longer need to lay them out one by one.
b77a127 - Provide utility access points to generate coordinates in a single method call without the Java bean style getter/setter.
0a9b2d2 - Handle more cases of fixed coordinates.
99a5a12 - Mark RingSets as placed when all their rings are placed.
86989b8 - Complex template for Ecteinascidin 743/770 etc.
ae84c56 - Critical Patch: stereochemistry must be assigned after molecule has been flipped! Otherwise it may be wrong.
308c34e - Protect against NPE when ringSystems.
e3c401d - Pass in fixed bonds to layout refiner.
2829ad5 - Some tweaks required for handling partially placed rings.
3af1363 - Some extra work needed for partially laid out ring systems, tidying up as reading.
dce48af - Don't seed a layout if part of the molecule is fixed by the caller.
752bd78 - Separate out layout seeding from continuation.
819a7f6 - Deprecate 'experimental' method that now serves little purpose.
3ec5fb4 - Only select orientation if afix is empty.
fb7e2a4 - We now have different cycle detection methods that should be deterministic. Also we sort the ring set a few lines later on by bond count.
3282f2c - Tidy up main entry method of SDG.
79a71b8 - New API points to allow partial layout of a molecule, we can allow the 'fixing' positions of atoms/bonds.
3b839da - Merge pull request #204 from cdk/patch/reactonsmarts
40eea14 - Allow optional map token in SMARTS parser.
4d2e5d3 - Tidy up how filters are applied in the SmartsPattern and include the new Atom-Atom Mapping filter.
9eef3b3 - A structure pattern filter for atom-atom maps.
f3fa698 - Correct map propagation when no right expression.
f2b70f9 - Copy across AAM idx to the resulting SMARTS Query Atoms.
11c5235 - Use StringBuilder not StringBuffer for non-threaded appends.
f275cea - Parse atom map into the AST.
cb1dc0e - Watch out for null component ids.
821d427 - Wire up Pattern implementation to accept reactions, a few basic tests are included for sanity checking.
19c8f91 - ReactionRole enum was moved from the SMARTS atom to the cdk-interfaces module.
e8baa77 - Fix imports.
4cae2a9 - Update component grouping predicate to consider reaction components as connected (even if they are ionic).
94b92dc - Conversion to/from inline reaction (molecules) that will allow us to easily implement matching.
729f5db - Allow stereo elements to be set on query atom containers.
de5fbcc - Rename query atom to reflect it's purpose.
f9a12b4 - Correct parsing and building of reaction SMARTS queries. Relatively simple adaption to existing parser.
f924784 - Merge pull request #203 from cdk/patch/sdfwriteinit
cf70611 - Make it explicit that a BufferedWriter can be passed in.
aabd1fb - Small fixup - didn't write the number of CTFileQueryBond and cleaner error handling.
e78d305 - Provide symbols when writing multiple group brackets - more concise representation, for example (H2O)8.
4e969aa - More robust bond order output for Molfile V2000.
6b1442b - Need to access the correct scale value from the model.
3f9fdab - Avoid repeated work when bending/stretching bonds to improve layout. Here we simply store a visit map and only bend/stretch a bond if it's the first pair it's seen in.
1577d14 - Improved multiple attachment point rendering by displaying number.
ae62d4d - Make generators thread-safe.
2d9a2b0 - Updated the copyright to say 2016.
fb48d9f - Update .travis.yml
f064df8 - Allow JavaDoc to built with java 8.
de1c1df - Development version 1.5.14-SNAPSHOT.
7e473ad