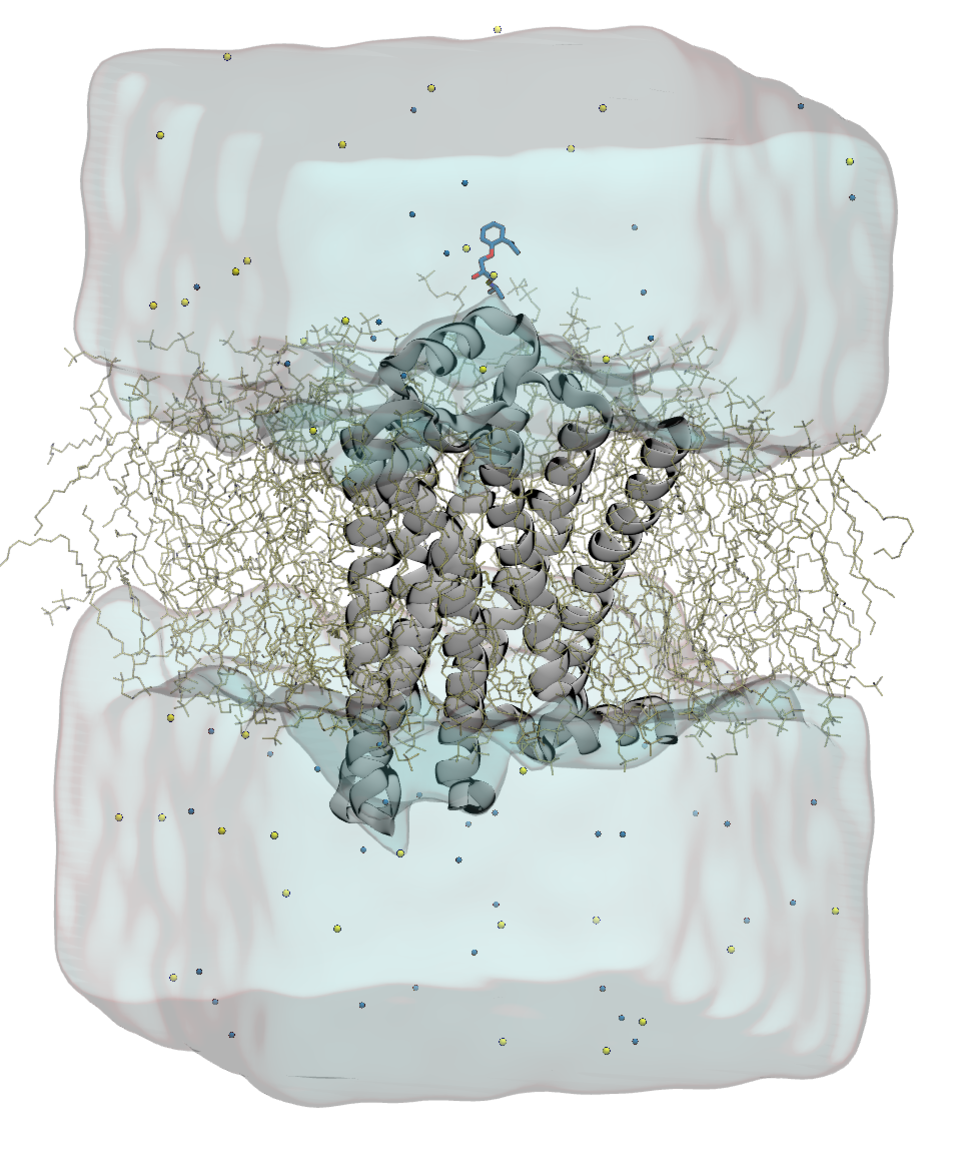
Dabble makes molecular dynamics system building easy!
No more messing with atom names or lipid membranes, Dabble does all the work for you. Supports multiple forcefields (CHARMM and AMBER), and simulation packages! Currently, that's CHARMM, AMBER, Desmond, and Anton!