/$$$$$$ /$$ /$$$$$$$ /$$$$$$
/$$__ $$ | $$ | $$__ $$ /$$__ $$
| $$ \ $$ /$$ /$$ /$$$$$$ /$$$$$$ | $$ \ $$| $$ \ $$
| $$$$$$$$| $$ | $$|_ $$_/ /$$__ $$| $$$$$$$ | $$$$$$$$
| $$__ $$| $$ | $$ | $$ | $$ \ $$| $$__ $$| $$__ $$
| $$ | $$| $$ | $$ | $$ /$$| $$ | $$| $$ \ $$| $$ | $$
| $$ | $$| $$$$$$/ | $$$$/| $$$$$$/| $$$$$$$/| $$ | $$
|__/ |__/ \______/ \___/ \______/ |_______/ |__/ |__/
Automated Bioinformatics Analysis
www.joshuachou.inkAn AI Agent for Fully Automated Multi-omic Analyses.
(Automated Bioinformatics Analysis via AutoBA)
Juexiao Zhou, Bin Zhang, Xiuying Chen, Haoyang Li, Xiaopeng Xu, Siyuan Chen, Wenjia He, Chencheng Xu, Xin Gao
King Abdullah University of Science and Technology, KAUST
demo.mp4
- [2024/01] Don't like the command line mode? Now we provide a new GUI and released the milestone stable version
v0.2.0🎉 - [2024/01] Updated JSON mode for gpt-3.5-turbo-1106, gpt-4-1106-preview, the output of these two models will be more stable
- [2024/01] Updated the support for ChatGPT-4 (gpt-4-32k-0613: Currently points to gpt-4-32k-0613, 32,768 tokens, Up to Sep 2021; gpt-4-1106-preview: GPT-4 Turbo, 128,000 tokens, Up to Apr 2023)
- [2024/01] Updated the support for ChatGPT-3.5 (gpt-3.5-turbo: openai chatgpt-3.5, 4,096 tokens and gpt-3.5-turbo-1106: openai chatgpt-3.5, 16,385 tokens)
- [2023/12] We added LLM support for the executor and the ACR module and released the milestone stable version
v0.1.1 - [2023/12] We provided the latest docker version to simplify the installation process.
- [2023/12] New feature: automated code repairing (ACR module) added, add llama2-chat backends.
- [2023/11] We updated the executor and released the latest stable version (v0.0.2) and are working on automatic error feedback and code fixing.
- [2023/10] We validated AutoBA on 40 conventional bioinformatics tasks and released our new pre-print at https://www.biorxiv.org/content/10.1101/2023.09.08.556814v2. More to come!
- [2023/09] We integrated codellama 7b-Instruct, 13b-Instruct, 34b-Instruct, now users can choose to use chatgpt or local llm as backends, we currently recommend using chatgpt because tests have found that codellama is not as effective as chatgpt for complex bioinformatics tasks.
- [2023/09] We are pleased to announce the official release of AutoBA's latest version
v0.0.1! 🎉🎉🎉
We're working hard to achieve more features, welcome to PRs!
- Automatic error feedback and code fixing
- Offer local LLMs (eg. code llama) as options for users
- Provide a docker version, simplify the installation process
- A UI-based YAML generator
- User Forum
- Pack into a conda package, simplify the installation process
- ...
# (mandatory) for basic functions
conda create -n abc python==3.10
conda activate abc
conda install -c anaconda yaml==0.2.5 -y
pip install openai==0.27.6 pyyaml==6.0
pip install transformers==4.34.0
git clone https://github.com/JoshuaChou2018/AutoBA.git
# (optional) for gui version
pip install gradio==4.14.0
# (optional) for local llm (llama2)
cd AutoBA/src/codellama-main
pip install -e .
## apply for a download link at https://ai.meta.com/resources/models-and-libraries/llama-downloads/
## download codellama model weights: 7b-Instruct,13b-Instruct,34b-Instruct
cd src/codellama-main
bash download.sh
## download llama2 model weights: 7B-chat,13B-chat,70B-chat
cd src/llama-main
bash download.sh
## download hf version model weights
git lfs install
cd src/codellama-main
git clone https://huggingface.co/codellama/CodeLlama-7b-Instruct-hf
git clone https://huggingface.co/codellama/CodeLlama-13b-Instruct-hf
git clone https://huggingface.co/codellama/CodeLlama-34b-Instruct-hf
# (optional) for features under development: the yaml generator UI
pip install plotly==5.14.1 dash==2.9.3 pandas==2.0.1 dash-mantine-components==0.12.1Please refer to https://docs.docker.com/engine/install to install Docker first.
# (mandatory) for basic functions
docker pull joshuachou666/autoba:cuda12.2.2-cudnn8-devel-ubuntu22.04-autoba0.1.2
docker run --rm --gpus all -it joshuachou666/autoba:cuda12.2.2-cudnn8-devel-ubuntu22.04-autoba0.1.2 /bin/bash
## Enter the shell in docker image
conda activate abc
cd AutoBAIf you get this error: could not select device driver "" with capabilities: [[gpu]], then run the following codes:
# (optional) for using GPU in docker
curl -s -L https://nvidia.github.io/nvidia-docker/gpgkey | \
sudo apt-key add -
distribution=$(. /etc/os-release;echo $ID$VERSION_ID)
curl -s -L https://nvidia.github.io/nvidia-docker/$distribution/nvidia-docker.list | \
sudo tee /etc/apt/sources.list.d/nvidia-docker.list
sudo apt-get update
sudo apt install -y nvidia-docker2
sudo systemctl daemon-reload
sudo systemctl restart dockerTry the previous codes again.
Coming soon..../example contains several examples for you to start.
Under ./example, config.yaml defines your files and goals. Defining data_list, output_dir and goal_description
in config.yaml is mandatory before running app.py. Absolute paths rather than relative paths are recommended for all file paths defined in config.yaml.
app.py run this file to start.
Run this command to start a simple example with chatgpt as backend (recommended).
python app.py --config ./examples/case1.1/config.yaml --openai YOUR_OPENAI_API --model gpt-4
Execute the code while generating it with ACR module loaded.
python app.py --config ./examples/case1.1/config.yaml --openai YOUR_OPENAI_API --model gpt-4 --execute True
Please note that this work uses the GPT-4 API and does not guarantee that GPT-3.5 will work properly in all cases.
or with local llm as backend (not recommended for the moment, in development and only for testing purposes)
python app.py --config ./examples/case1.1/config.yaml --model codellama-7bi
Run this command to start a GUI version of AutoBA.
python gui.py
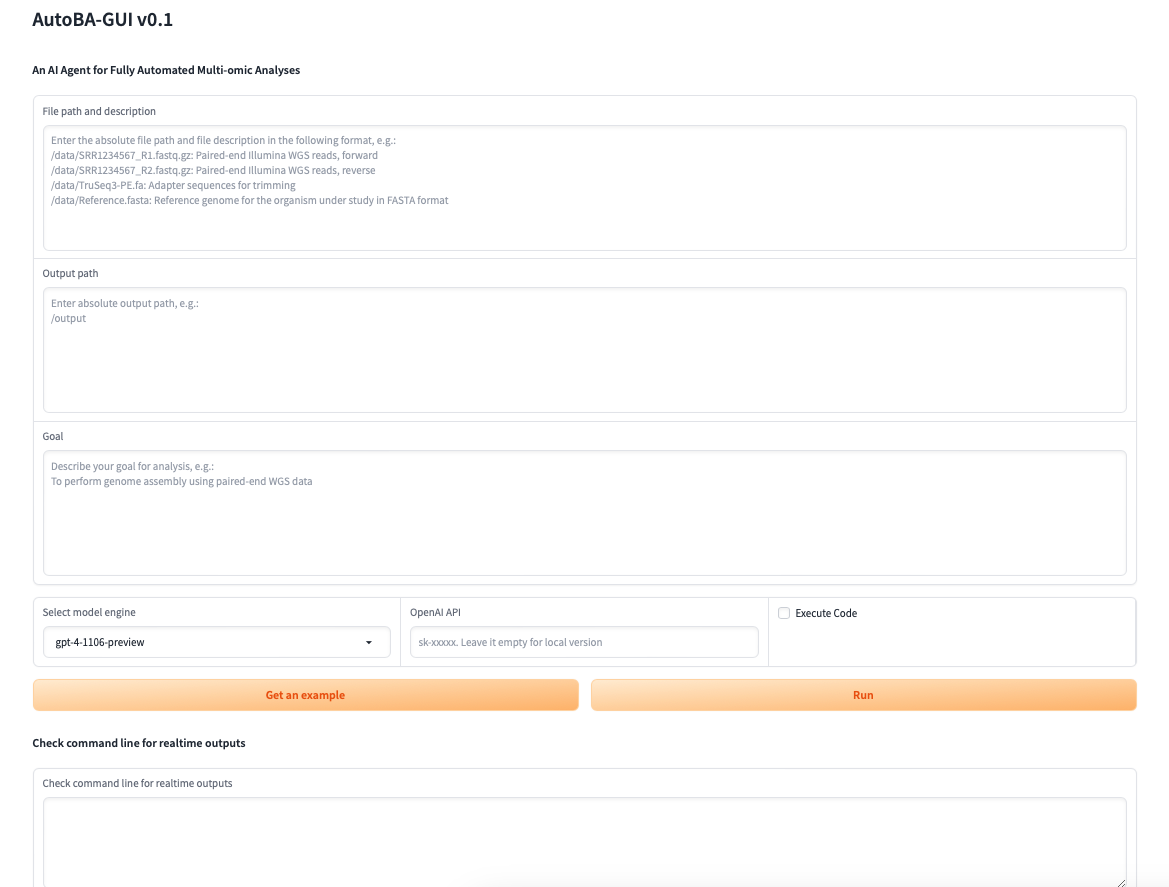
Dynamic Engine: dynamic update version
- gpt-3.5-turbo: Currently points to gpt-3.5-turbo-0613, 4,096 tokens, Up to Sep 2021
- gpt-4: Currently points to gpt-4-0613, 8,192 tokens, Up to Sep 2021 (default)
Fixed Engine: snapshot version
- gpt-3.5-turbo-1106: Updated GPT 3.5 Turbo, 16,385 tokens, Up to Sep 2021
- gpt-4-0613: Snapshot of gpt-4 from June 13th 2023 with improved function calling support, 8,192 tokens, Up to Sep 2021
- gpt-4-32k-0613: Snapshot of gpt-4-32k from June 13th 2023 with improved function calling support, 32,768 tokens, Up to Sep 2021
- gpt-4-1106-preview: GPT-4 Turbo, 128,000 tokens, Up to Apr 2023
- codellama-7bi: 7b-Instruct
- codellama-13bi: 13b-Instruct
- codellama-34bi: 34b-Instruct
- llama2-7bc: llama-2-7b-chat
- llama2-13bc: llama-2-13b-chat
- llama2-70bc: llama-2-70b-chat
These cases below may have different ID numbers as those cases in our paper.
Reference: https://pzweuj.github.io/worstpractice/site/C02_RNA-seq/01.prepare_data/
Design of config.yaml
data_list: [ './examples/case1.1/data/SRR1374921.fastq.gz: single-end mouse rna-seq reads, replicate 1 in LoGlu group',
'./examples/case1.1/data/SRR1374922.fastq.gz: single-end mouse rna-seq reads, replicate 2 in LoGlu group',
'./examples/case1.1/data/SRR1374923.fastq.gz: single-end mouse rna-seq reads, replicate 1 in HiGlu group',
'./examples/case1.1/data/SRR1374924.fastq.gz: single-end mouse rna-seq reads, replicate 2 in HiGlu group',
'./examples/case1.1/data/TruSeq3-SE.fa: trimming adapter',
'./examples/case1.1/data/mm39.fa: mouse mm39 genome fasta',
'./examples/case1.1/data/mm39.ncbiRefSeq.gtf: mouse mm39 genome annotation' ]
output_dir: './examples/case1.1/output'
goal_description: 'find the differentially expressed genes'wget -P data/ http://hgdownload.soe.ucsc.edu/goldenPath/mm39/bigZips/genes/mm39.ncbiRefSeq.gtf.gz
wget -P data/ http://hgdownload.soe.ucsc.edu/goldenPath/mm39/bigZips/mm39.fa.gz
gunzip data/mm39.ncbiRefSeq.gtf.gz
gunzip data/mm39.fa.gz
wget -P data/ ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR137/001/SRR1374921/SRR1374921.fastq.gz
wget -P data/ ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR137/002/SRR1374922/SRR1374922.fastq.gz
wget -P data/ ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR137/003/SRR1374923/SRR1374923.fastq.gz
wget -P data/ ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR137/004/SRR1374924/SRR1374924.fastq.gzpython app.py --config ./examples/case1.1/config.yaml --openai YOUR_OPENAI_API --model gpt-4
python app.py --config ./examples/case1.1/config.yaml --model codellama-7bi
python app.py --config ./examples/case1.1/config.yaml --model codellama-13bi
python app.py --config ./examples/case1.1/config.yaml --model codellama-34biReference: https://pzweuj.github.io/worstpractice/site/C02_RNA-seq/01.prepare_data/
Design of config.yaml
data_list: [ './examples/case1.2/data/SRR1374921.fastq.gz: single-end mouse rna-seq reads, replicate 1 in LoGlu group',
'./examples/case1.2/data/SRR1374922.fastq.gz: single-end mouse rna-seq reads, replicate 2 in LoGlu group',
'./examples/case1.2/data/SRR1374923.fastq.gz: single-end mouse rna-seq reads, replicate 1 in HiGlu group',
'./examples/case1.2/data/SRR1374924.fastq.gz: single-end mouse rna-seq reads, replicate 2 in HiGlu group',
'./examples/case1.2/data/TruSeq3-SE.fa: trimming adapter',
'./examples/case1.2/data/mm39.fa: mouse mm39 genome fasta',
'./examples/case1.2/data/mm39.ncbiRefSeq.gtf: mouse mm39 genome annotation' ]
output_dir: './examples/case1.2/output'
goal_description: 'Identify top5 down-regulated genes in HiGlu group'wget -P data/ http://hgdownload.soe.ucsc.edu/goldenPath/mm39/bigZips/genes/mm39.ncbiRefSeq.gtf.gz
wget -P data/ http://hgdownload.soe.ucsc.edu/goldenPath/mm39/bigZips/mm39.fa.gz
gunzip data/mm39.ncbiRefSeq.gtf.gz
gunzip data/mm39.fa.gz
wget -P data/ ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR137/001/SRR1374921/SRR1374921.fastq.gz
wget -P data/ ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR137/002/SRR1374922/SRR1374922.fastq.gz
wget -P data/ ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR137/003/SRR1374923/SRR1374923.fastq.gz
wget -P data/ ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR137/004/SRR1374924/SRR1374924.fastq.gzpython app.py --config ./examples/case1.2/config.yamlReference: https://github.com/STAR-Fusion/STAR-Fusion-Tutorial/wiki
Design of config.yaml
data_list: [ './examples/case1.3/data/rnaseq_1.fastq.gz: RNA-Seq read 1 data (left read)',
'./examples/case1.3/data/rnaseq_2.fastq.gz: RNA-Seq read 2 data (right read)',
'./examples/case1.3/data/CTAT_HumanFusionLib.mini.dat.gz: a small fusion annotation library',
'./examples/case1.3/data/minigenome.fa: small genome sequence consisting of ~750 genes.',
'./examples/case1.3/data/minigenome.gtf: transcript structure annotations for these genes' ]
output_dir: './examples/case1.3/output'
goal_description: 'Predict Fusion genes using STAR-Fusion'cd data
git clone https://github.com/STAR-Fusion/STAR-Fusion-Tutorial.git
mv STAR-Fusion-Tutorial/* .Reference: https://www.jianshu.com/p/e22a947e6c60
Design of config.yaml
data_list: [ './examples/case2.1/data/filtered_gene_bc_matrices/hg19: path to 10x mtx data',]
output_dir: './examples/case2.1/output'
goal_description: 'use scanpy to find the differentially expressed genes'wget -P data/ http://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz
cd data
tar zxvf pbmc3k_filtered_gene_bc_matrices.tar.gzReference: https://www.jianshu.com/p/e22a947e6c60
Design of config.yaml
data_list: [ './examples/case2.2/data/filtered_gene_bc_matrices/hg19: path to 10x mtx data',]
output_dir: './examples/case2.2/output'
goal_description: 'use scanpy to perform clustering and visualize the expression level of gene PPBP in the UMAP.'Reference: https://www.jianshu.com/p/e22a947e6c60
Design of config.yaml
data_list: [ './examples/case2.3/data/filtered_gene_bc_matrices/hg19: path to 10x mtx data',]
output_dir: './examples/case2.3/output'
goal_description: 'use scanpy to identify top5 marker genes'Reference: https://pzweuj.github.io/2018/08/22/chip-seq-workflow.html
Design of config.yaml
# 下载了5个样本,分别是Ring1B、cbx7、SUZ12、RYBP、IgGold。
for ((i=204; i<=208; i++)); \
do wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR620/SRR620$i/SRR620$i.fastq.gz; \
done
wget http://hgdownload.soe.ucsc.edu/goldenPath/mm39/bigZips/genes/mm39.ncbiRefSeq.gtf.gz
wget http://hgdownload.soe.ucsc.edu/goldenPath/mm39/bigZips/mm39.fa.gz
gunzip mm39.ncbiRefSeq.gtf.gz
gunzip mm39.fa.gzDesign of config.yaml
data_list: [ './examples/case3.1/data/SRR620204.fastq.gz: chip-seq data for Ring1B',
'./examples/case3.1/data/SRR620205.fastq.gz: chip-seq data for cbx7',
'./examples/case3.1/data/SRR620206.fastq.gz: chip-seq data for SUZ12',
'./examples/case3.1/data/SRR620208.fastq.gz: chip-seq data for IgGold',
'./examples/case3.1/data/mm39.ncbiRefSeq.gtf: genome annotations mouse',
'./examples/case3.1/data/mm39.fa: mouse genome' ]
output_dir: './examples/case3.1/output'
goal_description: 'call peaks for protein cbx7 with IgGold as control'Design of config.yaml
data_list: [ './examples/case3.2/data/SRR620204.fastq.gz: chip-seq data for Ring1B',
'./examples/case3.2/data/SRR620205.fastq.gz: chip-seq data for cbx7',
'./examples/case3.2/data/SRR620206.fastq.gz: chip-seq data for SUZ12',
'./examples/case3.2/data/SRR620208.fastq.gz: chip-seq data for IgGold',
'./examples/case3.2/data/mm39.ncbiRefSeq.gtf: genome annotations mouse',
'./examples/case3.2/data/mm39.fa: mouse genome' ]
output_dir: './examples/case3.2/output'
goal_description: 'Discover motifs within the peaks of protein SUZ12 with IgGold as control'Design of config.yaml
data_list: [ './examples/case3.3/data/SRR620204.fastq.gz: chip-seq data for Ring1B',
'./examples/case3.3/data/SRR620205.fastq.gz: chip-seq data for cbx7',
'./examples/case3.3/data/SRR620206.fastq.gz: chip-seq data for SUZ12',
'./examples/case3.3/data/SRR620208.fastq.gz: chip-seq data for IgGold',
'./examples/case3.3/data/mm39.ncbiRefSeq.gtf: genome annotations mouse',
'./examples/case3.3/data/mm39.fa: mouse genome' ]
output_dir: './examples/case3.3/output'
goal_description: 'perform functional enrichment for protein Ring1B, use protein IgGold as the control'Reference: https://squidpy.readthedocs.io/en/latest/notebooks/tutorials/tutorial_seqfish.html
Design of config.yaml
data_list: [ './examples/case4.1/data/slice1.h5ad: spatial transcriptomics data for slice 1 in AnnData format',]
output_dir: './examples/case4.1/output'
goal_description: 'use squidpy for neighborhood enrichment analysis'To use AutoBA in your case, please copy config.yaml to your destination and modify it accordingly.
Then you are ready to go. We welcome all developers to submit PR to upload your special cases under ./projects
We appreciate all contributions to improve AutoBA.
The main branch serves as the primary branch, while the development branch is dev.
Thank you for your unwavering support and enthusiasm, and let's work together to make AutoBA even more robust and powerful! If you want to contribute, please PR to dev. 💪
If you find this project useful in your research, please consider citing:
@article {Zhou2023.09.08.556814,
author = {Juexiao Zhou and Bin Zhang and Xiuying Chen and Haoyang Li and Xiaopeng Xu and Siyuan Chen and Wenjia He and Chencheng Xu and Xin Gao},
title = {An AI Agent for Fully Automated Multi-omic Analyses},
elocation-id = {2023.09.08.556814},
year = {2024},
doi = {10.1101/2023.09.08.556814},
publisher = {Cold Spring Harbor Laboratory},
abstract = {With the fast-growing and evolving omics data, the demand for streamlined and adaptable tools to handle the bioinformatics analysis continues to grow. In response to this need, we introduce Automated Bioinformatics Analysis (AutoBA), an autonomous AI agent designed explicitly for fully automated multi-omic analyses based on large language models. AutoBA simplifies the analytical process by requiring minimal user input while delivering detailed step-by-step plans for various bioinformatics tasks. Through rigorous validation by expert bioinformaticians, AutoBA{\textquoteright}s robustness and adaptability are affirmed across a diverse range of omics analysis cases, including whole genome/exome sequencing (WGS/WES), chromatin immunoprecipitation assays with sequencing (ChIP-seq), RNA sequencing (RNA-seq), single-cell RNA-seq, spatial transcriptomics and so on. AutoBA{\textquoteright}s unique capacity to self-design analysis processes based on input data variations further underscores its versatility. Compared with online bioinformatic services, AutoBA offers multiple LLM backends, with options for both online and local usage, prioritizing data security and user privacy. Moreover, different from the predefined pipeline, AutoBA has adaptability in sync with emerging bioinformatics tools. Overall, AutoBA represents an advanced and convenient tool, offering robustness and adaptability for conventional multi-omic analyses.Competing Interest StatementThe authors have declared no competing interest.},
URL = {https://www.biorxiv.org/content/early/2024/01/05/2023.09.08.556814},
eprint = {https://www.biorxiv.org/content/early/2024/01/05/2023.09.08.556814.full.pdf},
journal = {bioRxiv}
}
This project is released under the MIT license.