{kind=link}
Identification of fixed SNPs and InDels distinguishing homoplastic and non-homoplastic coregenome variants.
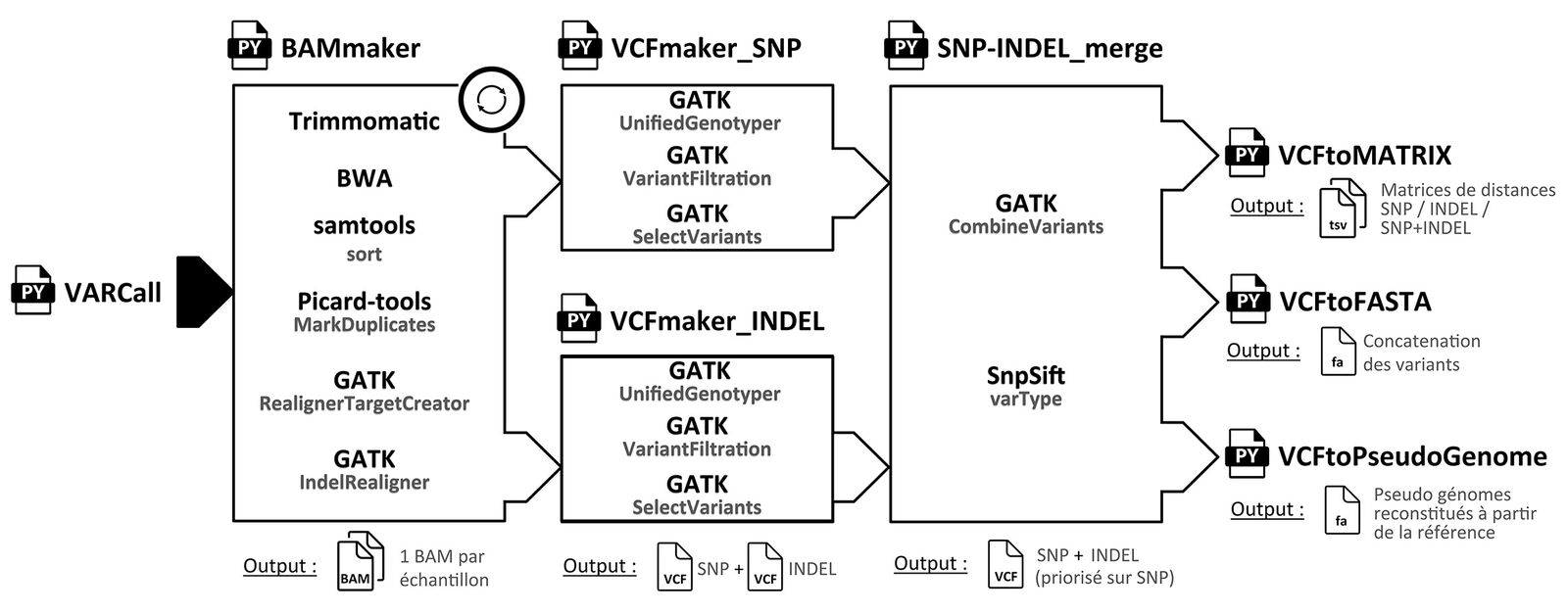
- Identify all comparisons of genome groups referring to all nodes,
- Select sensitive and specific variants,
- Define if these variants are involved the effect of homoplasy.
Gene ontology enrichment analysis based on hypergeometric tests, identifying genome groups and excluding obsolete and non-prokaryotic GO-terms.

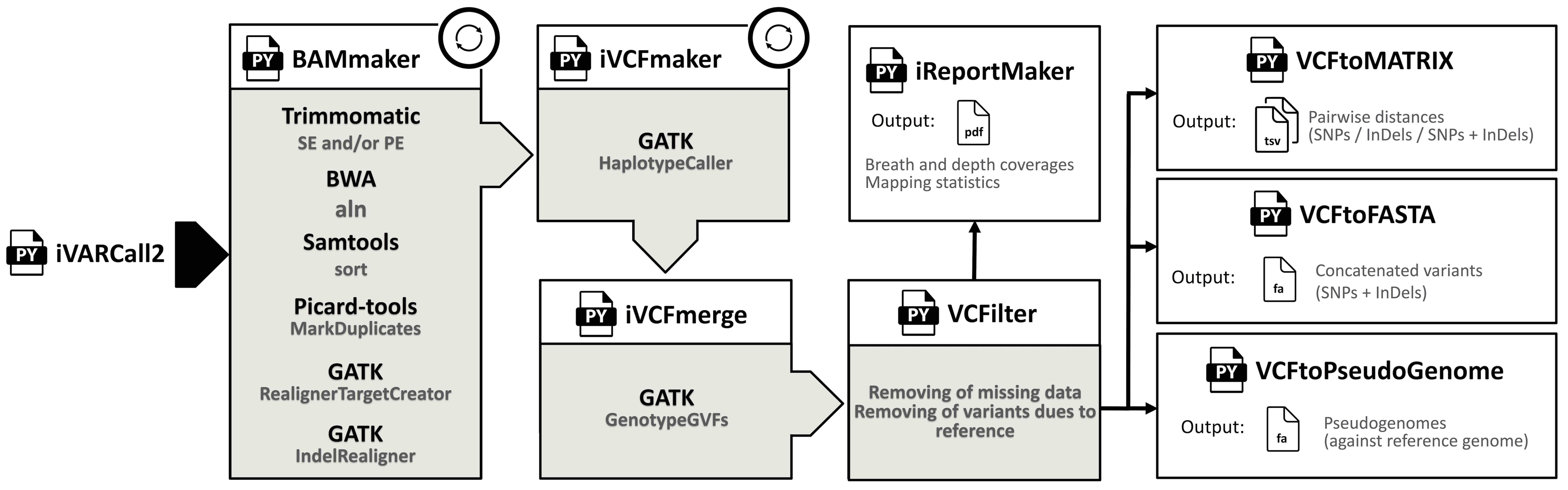
"Independant variant calling". Aims to perform a variant calling analysis from Illumina paired-end reads based on the GATK HaplotypeCaller algorithm. Each sample are processed independently and a g.vcf file is produce for each of them. This allows combination of several iVARCall2 results if the same reference genome is used.