{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
This repository gives you the code you'll need to kickstart a personal website that showcases your work as a software developer. And when you manage the code in a GitHub repository, it will automatically render a webpage with the owner's profile information, including a photo, bio, and repositories.
Your personal website is waiting to be personalized, though. It includes space to highlight your specific areas of interest in software development, like languages or industries. And it's standing by to publish your next great blog post.
It's all possible using the combination of Jekyll (for building your website), GitHub Pages (for hosting your website), and GitHub's API (for automatically populating your website with content).
You'll be making your own copy of the "personal website starter" repository so you have your own project to customize. A "fork" is a copy of a repository. So select "Fork" atop the github/personal-website repository.
Once you've found a home for your forked repository, it's yours. You're the owner, so you're ready to publish, if you wish.
If you want to manage your website in a local web development environment, you'll be using Ruby.
Once you've found a home for your forked repository, clone it.
Jekyll is a Ruby Gem that can be installed on most systems.
- Install a full Ruby development environment
- Install Jekyll and bundler gems
gem install jekyll bundler
- Change into your new directory
cd personal-website
- Install missing gems
bundle install
- Build the site and make it available on a local server
bundle exec jekyll serve
You should see something like:
Configuration file: /octocat/personal-website/_config.yml
Source: /octocat/personal-website
Destination: /octocat/_site
Incremental build: disabled. Enable with --incremental
Generating...
GitHub Metadata: No GitHub API authentication could be found. Some fields may be missing or have incorrect data.
done in 14.729 seconds.
Auto-regeneration: enabled for '/octocat/personal-website'
Server address: http://127.0.0.1:4000
Server running... press ctrl-c to stop.
Don't worry about the "No GitHub API authentication could be found" message. API authentication is only necessary if you intend to display more detailed metadata, like a branch name.
- Now browse to http://localhost:4000
When you host your personal website's code on GitHub, you get the support of free hosting through GitHub Pages.
The fastest approach is to rename your repository username.github.io, where username is your GitHub username (or organization name). Then, the next time you push any changes to your repository's master branch, they'll be accessible on the web at your username.github.io address.
If you want to use a custom domain, you'll want to add it to your repository's "Custom domain" settings on github.com. And then register and/or configure your domain with a DNS provider.
It's your website, and you control the source code. So you can customize everything, if you like. But we've provided a handful of quick customizations for you to consider as you get your website off the ground.
Most customizations can be done in a matter of seconds, by revising your repository's _config.yml file. Just remember to restart your local server each time you save new changes so your Jekyll-powered website rebuilds correctly:
- Shut down your server by entering the keyboard command CTRL+c
- Restart your server:
bundle exec jekyll serve
Your website will display in a two-column layout by default on larger-screen devices, with your photo, name, and basic information displayed in a left-aligned "sidebar." You can quickly switch to a "stacked" single-column layout by changing the line in your _config.yml file that reads layout: sidebar to layout: stacked.
Your website appears with a "light" white and gray background by default, with dark text. You can quickly switch to a "dark" background with white text by changing the line in your _config.yml file that reads style: light to style: dark.
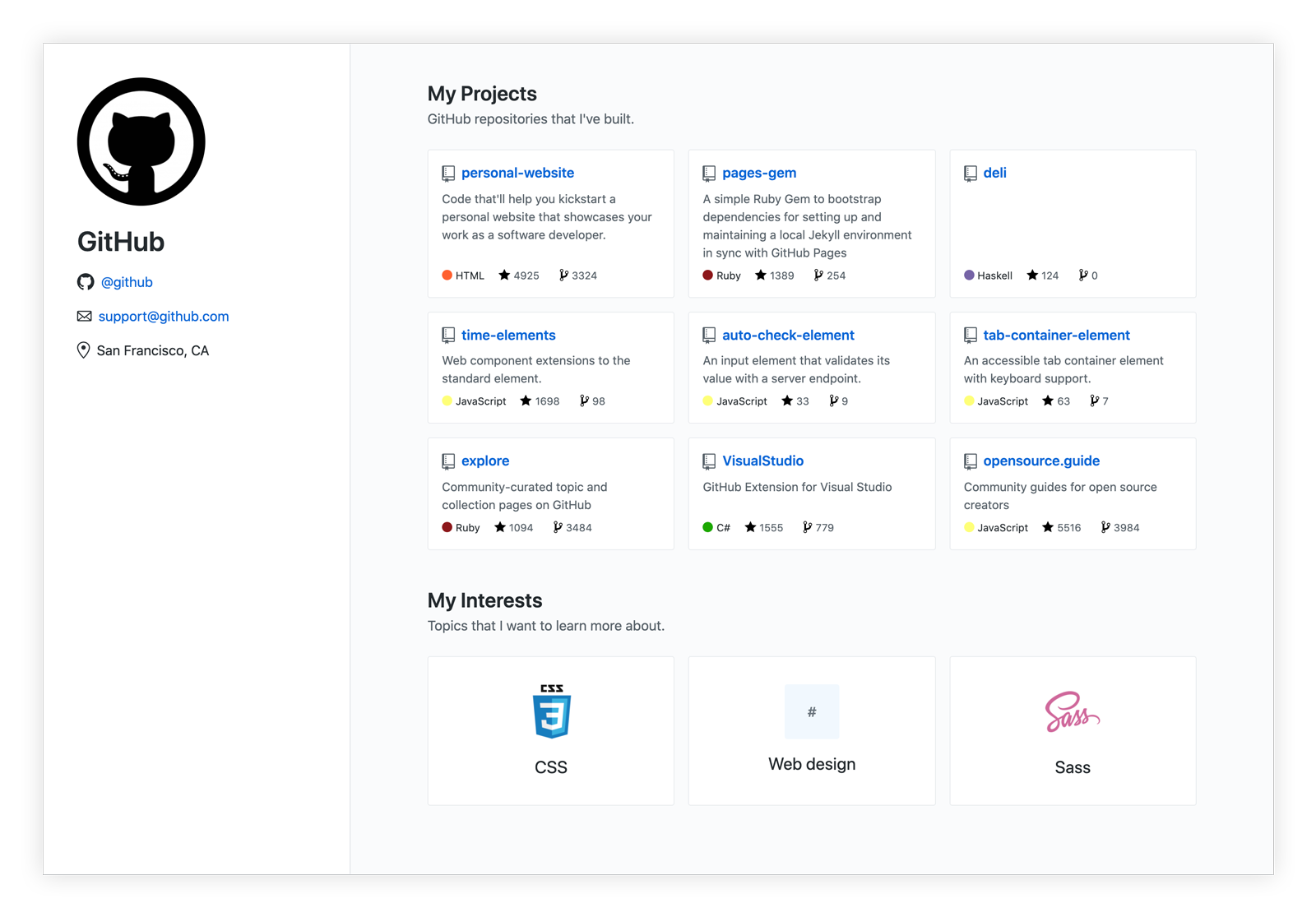
The "My Projects" section of your website is generated by default with your nine most recently "pushed" repositories. It also excludes repositories that you forked, by default. But each of these parameters can be quickly customized in your repository's _config.yml file, under the projects dictionary line.
Parameters include:
sort_by: The method by which repositories are sorted. Options includepushedandstars.limit: The maximum number of repositories that will be displayed in the "My Projects" section of your website. Out of the box, this number is set to9.exclude:forks: Whentrue, repositories you've forked will be excluded from the listing.projects: A list the repository names you want to exclude from the listing.
Your website comes pre-configured with three topics (e.g. "Web design" and "Sass") that appear in a section titled "My Interests." These are also stored in your repository's _config.yml file, where you can define each topic's name and two other optional details:
web_url: The web address you'd like to your topic to link to (e.g.https://github.com/topics/sass).image_url: The web address of an (ideally square) image that you'd like to appear with your topic.
Your website supports linking and sharing to social media services you're using, including Behance, Dribbble, Facebook, LinkedIn, Medium, Stack Overflow, Twitter, and YouTube. To identify the services you use:
- Edit your repository's
_config.ymlfile. - Edit the
social_mediadictionary line, and represent the services you like in a simplekey: valueform:
social_media:
behance: your_username
dribbble: your_username
facebook: your_username
hackerrank: your_username
instagram: your_username
keybase: your_username
linkedin: your_username
medium: your_username
stackoverflow: your_user_id
telegram: your_username
twitter: your_username
unsplash: your_username
vk: your_username
website: http://your_website_url
youtube: your_username
Links to your profile for each of the services you define will appear in the <header> of your website, appended to your bio. And if those services support sharing, any blog posts that you publish will include links to share that post using each social media service.
Note: This feature is supported by two files in your repository:
/_data/social_media.yml: Defines each of the supported services, including variable name, display name, URL path, and SVG icon./_includes/social_media_share_url.html: Outputs the share URL required for any of the supported social media services that support sharing URLs.
If you're interested in adding a social media service that's not already supported in this repo, you can edit these two files to build that support.
To add a page to your website (e.g. detailed resume):
- Create a new
.htmlor.mdfile at the root of your repository. - Give it a filename that you want to be used in the page's URL (e.g.
http://yoursite.dev/filename). - At the start of your file, include the following front matter:
---
layout: default
---
To add a blog post to your website:
- Create a new
.mdfile in your repository's/_posts/directory. - Give it a filename using the following format:
YEAR-MONTH-DAY-title.MARKUP
- At the start of your file, include the following front matter:
---
title: "The title of my blog post"
---
Your website comes with a placeholder blog post that you can reference. Notably, its front matter declares published as false, so that it won't appear on your website.
While you can define a layout in the front matter, your website is pre-configured to assign the post layout to all of the posts in your /_posts/ directory. So you don't have to declare that in your posts.
Jekyll's conventions for authoring and managing blog posts is very flexible. You can learn more in Jekyll's documentation for "Posts."
To give you a sound foundation to start your personal website, your repository includes a handful of "includes" -- dynamic .html files that are re-used throughout your website. They're all stored in the /_includes/ directory.
There are the usual suspects, like header.html and footer.html. But there are few more worth pointing out:
interests.html: A heading and dynamic list of "My Interests," which is populated with the topics you list in your_config.yml.masthead.html: A collection of your avatar, name, bio, and other metadata that's displayed prominently on all your webpages to help identify what the website is about.post-card.html: A compact, summarized presentation of a blog post, re-used to display a listing of your latest blog posts.projects.html: A heading and dynamic list of "My Projects," which is populated with a listing of your newest GitHub repositories.repo-card.html: A compact, summarized presentation of a repository, re-used to display a listing of your GitHub repositories.thoughts.html: A heading and dynamic list of "My Thoughts," which is populated with a listing of your latest blog posts.topic-card.html: A compact, summarized presentation of a topic (defined in your_config.yml), re-used to display a listing of your interests.
Your repository comes with three layouts:
- default: Not used by any of the built-in pages or posts, but useful for any new pages you create.
- home: Used by your
index.htmlhomepage to display listings of your projects, interests, and (optionally) your blog posts. - post: Used by default by the posts in your
/_posts/directory.
Jekyll's convention for defining layouts is very flexible. You can learn more about customizing your layouts in the Jekyll "Layouts" docs.
Your website is pre-configured to use GitHub's very flexible CSS framework called "Primer,". It's currently referenced within your styles.scss file, using the CSS import at-rule:
@import url('https://unpkg.com/primer/build/build.css');
You are, of course, welcome to remove it or replace it with another framework. Just bear in mind that the HTML that your website came pre-packaged with references multiple Primer "utility classes" to define things like column widths, margins, and background colors.
You also have the option to add on to and extend Primer's styles by adding custom CSS to your /assets/styles.scss Sass stylesheet. By editing this file, you can customize your website's color scheme, typography, and more.
The theme is available as open source under the terms of the MIT License.