This repository contains all of the code and analyses associated with a phylogenetic analysis of siphonophores. This work is presented in the following paper:
Munro C, Siebert S, Zapata F, Howison M, Damian Serrano A, Church SH, Goetz FE, Pugh PR, Haddock SHD, Dunn CW. (2018) Improved phylogenetic resolution within Siphonophora (Cnidaria) with implications for trait evolution. Molecular Phylogenetics and evolution.https://doi.org/10.1016/j.ympev.2018.06.030
This paper is also available as a biorxiv pre-print:
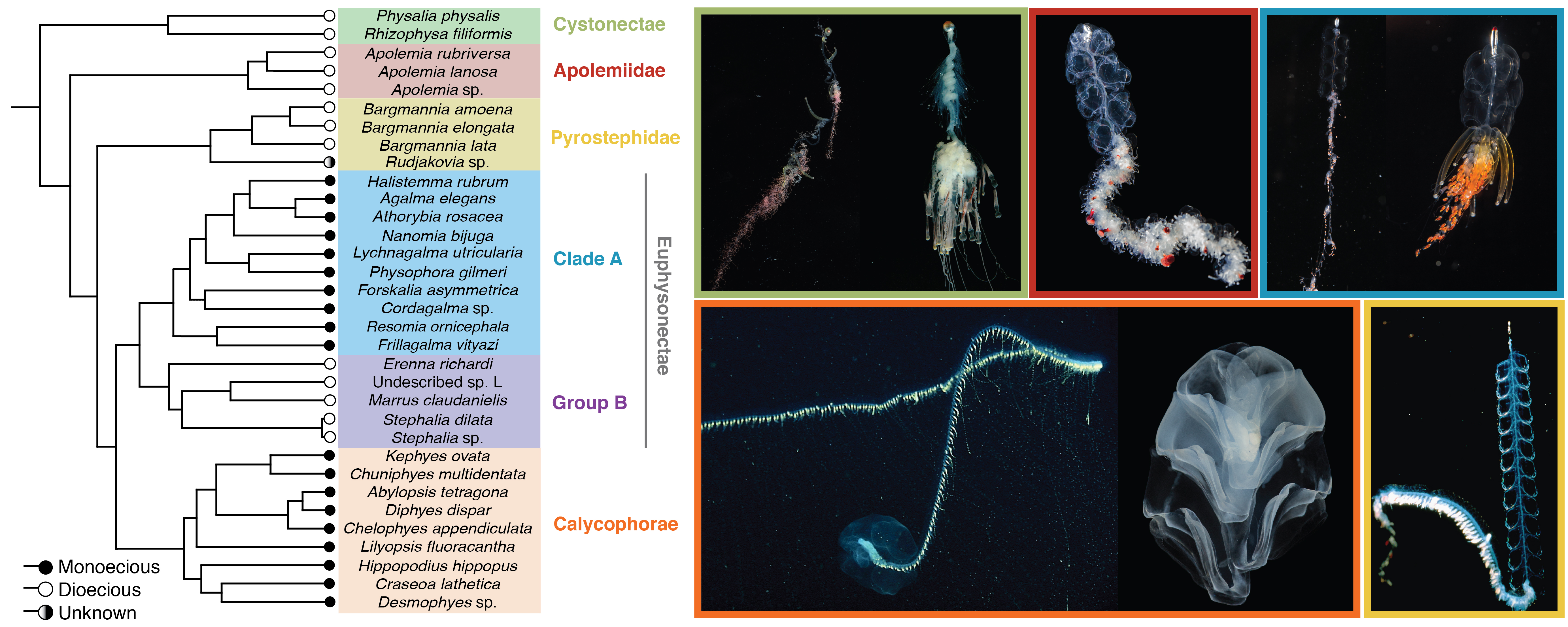
See figures for original figures used in this manuscript.
The repository includes a number of files needed to recreate these analyses. Sequences will need to be downloaded from the SRA numbers listed in supplementary_info/ (Supplementary_data_1_Specimen_data.csv) or from public databases (JGI, NCBI-EST etc). The respository also includes the following folders:
-
agalma/Code used to build the data matrix with Agalma v 1.0.0 & also downstream phylogenetic analyses. It also includes a folderSRA/that includes all of the information needed to modify the raw SRA files of some species for use in the analysis -
character_coding/Character coding files for the species considered in this manuscript -
sowh/Code used to carry out the SOWH tests -
manuscript.rmdSource code for the manuscript itself -
manuscript.pdfThe formatted manuscript