Bug in vcf.utils.walk_together(..) even when .vcfs are sorted correctly #140
Comments
|
See #20 |
|
Just as a side note that I hope can be useful: We've also just figured this out in our team (@ffinfo has the credit here). We've been trying to isolate a weird behavior in one of our scripts and it came down to this. What we found out was that the ordering follows a regular string ordering due to the way In short: So one way custom ordering can be implemented is if we allow the Alternatively, we can also allow the custom function to be plugged into |
- Added 'vcf_record_sort_key' to allow user to specify arbitrary chromosome ordering. - Fixed issue #140 by making sure to emit all records from the current chromosome before moving on to the next one. This takes care of the problem in most typical cases (eg. when all files have records for all contigs), but not in some edge cases, in which case the 'vcf_record_sort_key' arg can be used to fully solve the problem by explicitly defining the chromosome order.
|
Merged #143 |
|
@datagram, ah ~ I didn't realize this was already written as a pull request. It looks ok, and seeing that it's been merged is good I think :). |
- Added 'vcf_record_sort_key' to allow user to specify arbitrary chromosome ordering. - Fixed issue jamescasbon#140 by making sure to emit all records from the current chromosome before moving on to the next one. This takes care of the problem in most typical cases (eg. when all files have records for all contigs), but not in some edge cases, in which case the 'vcf_record_sort_key' arg can be used to fully solve the problem by explicitly defining the chromosome order.
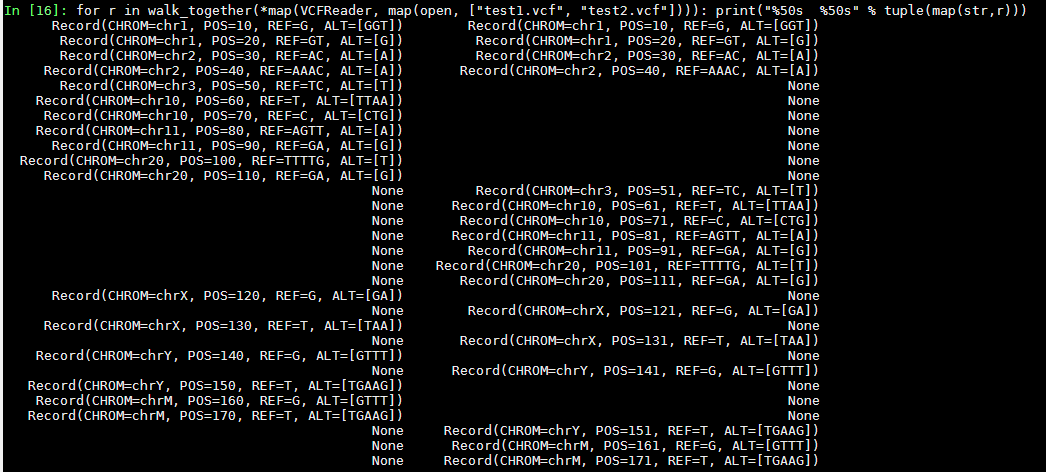
Thanks for creating this library and continuing to improve it. I just came across the following issue. walk_together uses a purely alphabetical ordering of chromosome names and so isn't aware that chr9 < chr10 or that chrY < chrM. As a result, it almost always produces unexpected behavior which, even when discovered, isn't useful in any situations I'm aware of, so I'm posting it as a bug.
For example, if you pass it 2 vcf files that have 4 variants each:
file1.vcf:
chr9 100 ...
chr9 200 ...
chr10 300 ...
chr10 400 ...
file2.vcf:
chr9 100 ...
chr9 210 ...
chr10 300 ...
chr10 410 ...
it will emit:
(file1-record1, file2-record2) # expected
(file1-record2, None) # expected
(file1-record3, None) # expecting (None, file2-record2) instead
(file1-record4, None) # expecting (file1-record3, file2-record3) instead
(None, file2-record2)
(None, file2-record3)
(None, file2-record4)
I'm not sure about the best way to make this function aware of the user's desired contig ordering, but as a simple solution it could take a list of contigs in their expected order (and then only emit records in those contigs).
Also, I think it would be nice if walk_together could raise an exception if any of the VCFs have conflicting contig orders.
-Ben
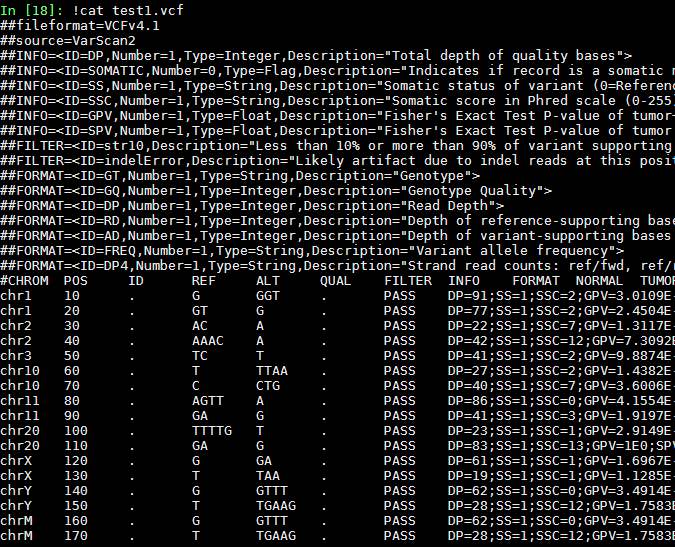
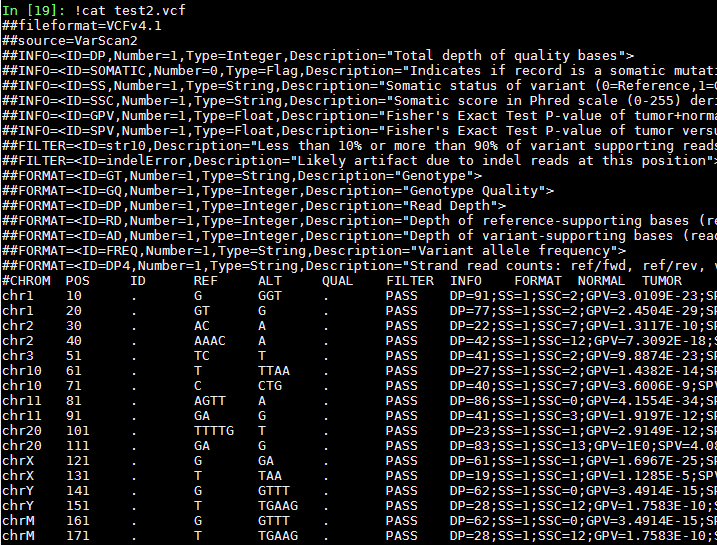
Ps. Below is a demonstration with 2 .vcf files with 17 variants each:
test1.vcf:
test2.vcf:
results for walk_together(test1, test2):
The text was updated successfully, but these errors were encountered: