Home
 |
 |
|---|
The version in this repository is for use on NIH HPC systems only. A version for generic Linux systems is coming out VERY SOON but the meantime, feel free to check out the JAMS preprint.
JAMS is a package in R for processing "microbiology" sequencing data. This means you can use it, for instance, for the following applications:
- Shotgun metagenomic data
- Shotgun metatranscriptomic data
- Whole genome sequencing of bacterial isolates
- Bacterial isolate transcriptomics
To get useful results from microbiological sequencing, JAMS splits the two phases of an analysis which are:
-
Determining the quantity and qualities of both taxonomic and functional information within a sample;
-
Determining statistical differences between samples.
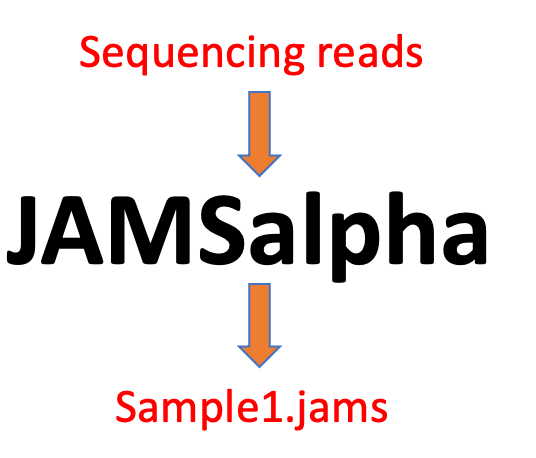
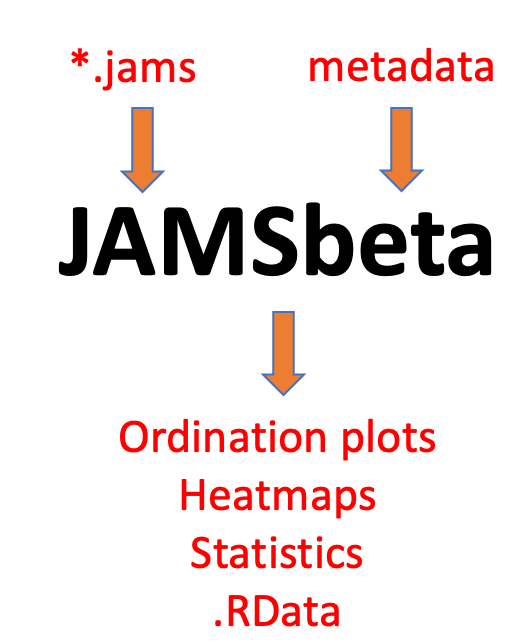
These two phases are dealt with with two separate command-line executables which run on Linux systems with R. JAMSalpha takes as input either sequencing reads or contigs for a single sample and yields, in addition to a report, a single file (samplename.jams) pertaining to that sample. Statistical analysis between samples can then be achieved with using the JAMSbeta executable to generate heatmaps, relative abundance plots, ordination plots and comparative alpha diversity plots, among others.
| JAMSalpha | JAMSbeta |
|---|---|
 |
 |