Standalone Code: Input File
RustBCA is configured through a TOML-format input file, input.toml. An example input file is located at the bottom of each of the example pages: 2D Geometry, Layered Targets, Distributions, and Multiple Interaction Potentials, and Spherical Geometry; there are also some additional examples in the repository under rustbca/examples/. The code can be run from the command line by simply calling ./rustbca on UNIX systems or rustBCA.exe on Windows; in this case, RustBCA will default to input.toml as the input file. Alternatively, one can run the code through cargo using cargo run --release, or to run with optional features, cargo run --release --features cpr_rootfinder,hdf5_input,distributions,no_list_output.
RustBCA accepts up to two command line arguments that allow the user to 1) specify an input file named something other than input.toml and 2) specify the type of geometry. If one gives RustBCA no command line arguments, it assumes a 2D Mesh input file and uses input.toml. One command line argument lets the user specify the input file: ./RustBCA input_file.toml. Using two command line arguments allows the user to specify both the input file and the geometry type (current options: 0D, 1D, 2D, TRIMESH, SPHERE, and, on the dev branch, HOMOGENEOUS2D): ./RustBCA 1D input_file_1D.toml.
NOTE: When writing floats by hand, you must include a terminal zero. toml-rs does not interpret terminal decimal points as valid floating point numbers. E.g., you must use 10.0 instead of 10. when writing input files by hand. This is not an issue when writing input files with the toml Python library, so long as the specific numpy encoder is used (e.g., toml.dump('filename.toml', encoder=toml.TomlNumpyEncoder()).
-
name (str): output file name prefix.
-
track_trajectories (optional, bool): track particle trajectories and output them to files [name]trajectory_data.output and [name]trajectories.output. Defaults to
false. -
track_recoils (optional, bool): track recoils generated by ion collisions with target atoms. Must be
trueto simulate sputtering. Defaults totrue. -
track_recoil_trajectories (optional, bool): include atomic recoils in trajectory output files.
track_recoilsmust be on. Defaults tofalse. -
track_displacements (optional, bool): whether or not to produce [name]displacements.output file. This output file lists all target atoms displaced with transferred energy T >
Ed. Defaults tofalse. -
track_energy_losses (optional, bool): whether to produce [name]energy_loss.output file. Defaults to
false. -
write_buffer_size (optional, int): size of streaming write buffer in bytes. Defaults to 8 KiB.
-
weak_collision_order (optional, int): number of weak collisions to simulate every binary collision step to emulate many-body effects. Weak collision partners are located in the plane normal to the particle trajectory, at random azimuthal angles, within annuli of radii
sqrt(k + [0, 1))*p_max, where k is the weak collision order and p_max is the maximum impact parameter. Defaults to 0. Warning: turning on weak collisions may result in erroneous results when Z1 >> Z2. -
suppress_deep_recoils (optional, bool): use Eckstein's empirical deep recoil suppression routine to speed up high energy sputtering simulations. See [1] for more details. Defaults to
false. -
high_energy_free_flight_paths (optional, bool): Use TRIM-style high-energy free flight paths between collisions to speed up high-energy simulations. Can only be used with Biersack-Varelas interpolated electronic stopping and without weak collisions. See [2] for more details. Defaults to
false. -
num_threads (optional, int): number of threads. Defaults to 1.
-
num_chunks (optional, int): number of chunks to split incident particles into. Each chunk is processed in parallel and written to disk before proceeding to the next one. Increase this option if RustBCA is using excessive system memory for large simulations. Defaults to 1.
-
use_hdf5 (optional, bool): whether or not to use hdf5 input for incident particles. Must build with
--features hdf5_input. Defaults tofalse. -
electronic_stopping_mode (optional, enum): mode of electronic stopping. Defaults to
"LOW_ENERGY_NONLOCAL"; must switch to interpolated for accuracy for incident energies above ~25 keV/amu. One of:-
"INTERPOLATED": Biersack-Varelas interpolation[2] (~10 eV - ~1 GeV) -
"LOW_ENERGY_NONLOCAL": Lindhard-Scharff nonlocal[3] (< 25keV/nucleon) -
"LOW_ENERGY_LOCAL": Oen-Robinson local[4] (<25 keV/nucleon) -
"LOW_ENERGY_EQUIPARTITION": Equipartition[5]
-
-
mean_free_path_model (optional, enum): how to determine mean-free-path distance. Defaults to
"LIQUID".-
"LIQUID": Liquid/Amorphous Solid: constant mean-free-path
-
"GASEOUS": Gaseous: exponentially distributed mean-free-path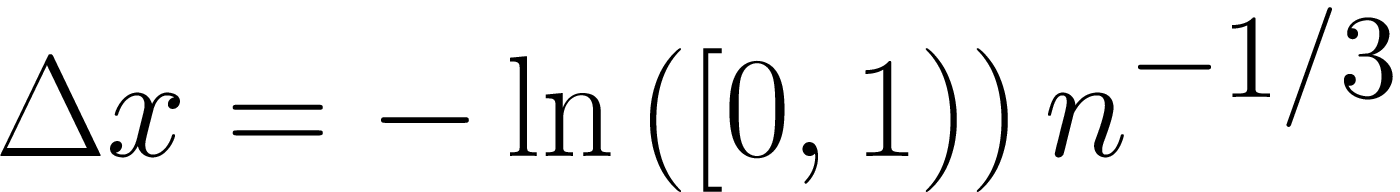
-
"THRESHOLD{density = (float)}": Exponentially distributed mean-free-path for geometry elements with a total number density less than the threshold density, constant mean-free-path otherwise; allows for the simulation of targets with some gaseous and some solid geometry elements.
-
-
interaction_potential NXN array(optional, enum), where N is the number of distinct interactions: atomic interaction potential. Defaults to
"KR-C".-
"MOLIERE": Moliere[6] -
"KR_C": Kr-C[7] -
"ZBL": ZBL[8] -
"LENZ_JENSEN": Lenz-Jensen[9] -
{"LENNARD_JONES_12_6"={sigma = 1E-10, epsilon = 1.6E-19}}, Lennard Jones 12-6 potential with sigma and epsilon adjustable parameters
-
{"LENNARD_JONES_65_6"={sigma = 1E-10, epsilon = 1.6E-19}}Lennard-Jones 6.5-6 potential with sigma and epsilon adjustable parameters
-
{"MORSE"={D = 1.6E-19, alpha = 1E10, r0 = 1E-10}}Morse potential with D, alpha and r0 adjustable parameters
-
-
scattering_integral NXN array(optional, enum), where N is the number of distinct interactions: scattering angle calculation method. Defaults to
"MENDENHALL-WELLER".-
"MENDENHALL_WELLER": Mendenhall-Weller 4-point Gauss-Lobatto quadrature[10]. Only usable with purely repulsive potentials. -
"MAGIC": MAGIC[11] algorithm. Only usable with purely repulsive potentials. -
{"GAUSS_MEHLER"={n_points = 10}}: Gauss-Mehler quadrature with n_points of integration -
"GAUSS_LEGENDRE": Gauss-Legendre 5-point quadrature
-
-
root_finder NXN array(optional, enum), where N is the number of distinct interactions: Rootfinder for distance of closest approach determination. See the rcpr crate for explanation of CPR options. Must build with
--features cpr_rootfinderto use CPR and Polynomial rootfinders.-
"DEFAULTNEWTON"default Newton-Raphson with 100 max iterations and a tolerance of 1E-3. Only usable with purely repulsive potentials. -
{"NEWTON"={max_iterations = 100, tolerance = 1E-6}}with adjustable tolerance and max iterations. Only usable with purely repulsive potentials. -
{"POLYNOMIAL"={complex_threshold=1E-12}}with adjustable threshold of imaginary component to ignore. Only usable with interaction potentials with a valid polynomial form. -
{"CPR"={n0=2, nmax=100, epsilon=1E-6, complex_threshold=1E-12, truncation_threshold=1E-12, far_from_zero=1E6, interval_limit=1E-16, derivative_free=false}}see the rcpr crate for explanation of CPR parameters.derivative_freeoption toggles between Newton-polishing (which requires a derivative of the doca function) and secant-polishing of roots.
-
The following options in the [options] block are used to customize histogram output if the code is built with --features distributions:
- energy_min (float): minimum energy value for binned distribution output
- energy_max (float): maximum energy value for binned distribution output
- energy_num (int): number of energy bins
- angle_min (float): minimum angle for binned distribution output
- angle_max (float): maximum angle for binned distribution output
- angle_num (float): number of angle bins
- x_min (float): minimum x value for binned distribution output
- y_min (float): minimum y value for binned distribution output
- z_min (float): minimum z value for binned distribution output
- x_max (float): maximum x value for binned distribution output
- y_max (float): maximum y value for binned distribution output
- z_max (float): maximum z value for binned distribution output
- x_num (int): number of x bins
- y_num (int): number of y bins
- z_num (int): number of z bins
-
energy_unit (string): unit of energy quantities in material input section. Can be one of: J, EV, KEV, MEV, or an arbitrary float, e.g. "1e-6", for a value in Joules
-
mass_unit (string): unit of mass quantities in material input section. Can be one of: AMU, KG, or an arbitrary float, e.g. "1e-6", for a value in kilograms
-
Eb (list(float)): Bulk binding energy of each species in the material.
-
Es (list(float)): Surface binding energy of each species in the material.
-
Ec (list(float)): Cutoff energy for each species in the material; i.e., the energy below which particles are considered stopped.
-
Ed (optional, list(float)): Displacement energy. Used to filter list output of displacement-producing collisions; defaults to 0.0.
-
Z (list(int)): Atomic number of each species in the material.
-
m (list(float)): Atomic mass of each species in the material.
-
interaction_index (optional, list(int)): index of interaction matrix to use for each species. Defaults to
[0], which means all interactions use the same potential that occupies i=0, j=0 of the interaction index matrix. -
surface_binding_model (optional, enum): which surface binding model to use. One of:
"PLANAR","ISOTROPIC", with a single inner arugment,calculation, which is one of"TARGET","INDIVIDUAL","AVERAGE". PLANAR refracts particles through a planar surface binding potential; ISOTROPIC does not cause particles to refract. INDIVIDUAL uses the individual surface binding energies for each particle regardless of composition. Alternatively, one can supply only the inner argument to default to the PLANAR surface binding model. TARGET uses the concentration-weighted linear combination of target surface binding energies, except when either the material or particle surface binding energy is zero, in which case it is zero. AVERAGE uses the average of the previous linear combination and the particle surface binding energy, except when the surface binding energy of either the material or target is zero, in which case it is zero. Defaults to"PLANAR"and"TARGET". -
bulk_binding_model (optional, enum): which bulk binding model to use. One of:
"INDIVIDUAL","AVERAGE". INDIVIDUAL uses the individual bulk binding energies for each particle regardless of composition. AVERAGE uses the concentration-weighted average of the material's bulk binding energies, except when the bulk binding energy of the particle is zero, in which case it is zero. Defaults to"AVERAGE".

The surface binding energy is the interaction energy of a particle with the surface, modeled as a planar, step potential in rustbca. When a particle approaches the energy barrier from the outside, if the surface binding energy is nonzero, it is accelerated slightly towards the surface, and refracted towards steeper angles of incidence. If a particle approaches the energy barrier from the material side, it will reflect back into the material if the perpendicular component of the kinetic energy is less than the surface binding energy. If it is greater, it will be refracted towards shallower exit angles. Particles outside the energy barrier will not be stopped until they either reenter the material or leave the simulation boundary. In other words, particles can only be deposited within the energy barrier, but may be deposited between the energy barrier and the material, analogous to deposition on the surface.

The bulk binding energy is the average energy required to remove a material atom from the lattice. In the BCA, it is applied as a one-time energy loss to atoms moved from their original locations in the material. NOTE: this is distinct from the displacement energy, which is the energy transfer necessary to create a stable Frenkel Pair. Bulk binding energies are typically ~3eV for metals.
- length_unit (string): unit of length quantities in particle input section. Can be one of: ANGSTROM, NM, MICRON, CM, MM, M, or an arbitrary float, e.g. "1e-6", for a value in meters. NOTE: if using an arbitrary float, it must be enclosed in quotation marks to be correctly parsed.
- energy_unit (string): unit of energy quantities in particle input section. Can be one of: EV, J, or an arbitrary float, e.g. "1e-6", for a value in Joules
- mass_unit (string): unit of mass quantities in particle input section. Can be one of: AMU, KG, or an arbitrary float, e.g. "1e-6", for a value in kilograms
- N (list(int)): number of incident ions to simulate
- m (list(float)): Atomic mass of incident ions
- Z (list(int)): Atomic number of incident ions
- E (list(float)): Initial energy of incident ions. Alternatively, one can use one of the following random distributions for the initial energy:
- Normal distribution:
{mean=f64, std=f64} - Uniform distribution:
{min=f64, max=f64}
- Ec (list(float)): Cutoff energy of incident ions
- Es (list(float)): Surface binding energy of incident ions
-
interaction_index (optional, list(int)): index of interaction matrix to use for this species. Defaults to
[0], which means all particles use the same interaction potential. - pos (list((float, float, float)): Initial x, y, and z of incident ions. Alternatively, one can use one of the following random distributions for any of the particle positions:
- Normal distribution: {mean=f64, std=f64} EXAMPLE:
{mean=1.0, std=1.0} - Uniform distribution: {min=f64, max=f64} EXAMPLE:
{min=0.0, max=5.0}
- dir (list((float, float, float)): Initial x-, y-, and z-direction of incident ions. Note: x-direction cannot be equal to zero or exactly equal to 1.0 to avoid gimbal lock. On initialization, dir is normalized to a unit vector, so there must be a non-zero y or z component. Alternatively, one can use one of the following random distributions for any of the particle positions:
- Normal distribution:
{mean=f64, std=f64} - Uniform distribution:
{min=f64, max=f64}
- particle_input_filename (string): name of HDF5 input file (optional)
Sphere is a homoegeneous spherical target with a user-defined radius with a center at the origin. For Sphere, the energy barrier thickness is set automatically.
- length_unit (string): unit of length quantities in Mesh2D input section. Can be one of ANGSTROM, NM, MICRON, CM, MM, M, or an arbitrary float, e.g. "1e-6", for a value in meters
- electronic_stopping_correction_factor (float): correction factor for electronic stopping. Default 1.0
- densities (list(float)): number densities for each species in material_parameters that make up the homogeneous target.
- radius (float): the sphere's radius.
Mesh 0D is a homogeneous target that begins at x = 0 with infinite width in y and z and infinite depth in x. For Mesh0D, the energy barrier thickness is set automatically.
- length_unit (string): unit of length quantities in Mesh2D input section. Can be one of ANGSTROM, NM, MICRON, CM, MM, M, or an arbitrary float, e.g. "1e-6", for a value in meters
- electronic_stopping_correction_factor (float): correction factor for electronic stopping. Default 1.0
- densities (list(float)): number densities for each species in material_parameters that make up the homogeneous target.
Mesh 1D is an inhomogeneous target that begins at x = 0 with infinite width in y and z and finite depth in x. For Mesh1D, the energy barrier thickness is set automatically. User provides individual layer thicknesses; the maximum depth is the sum of all the layer thicknesses.
- length_unit (string): unit of length quantities in Mesh2D input section. Can be one of ANGSTROM, NM, MICRON, CM, MM, M, or an arbitrary float, e.g. "1e-6", for a value in meters
- electronic_stopping_correction_factors (list(float)): correction factor for electronic stopping in each layer. Default 1.0
- layer_thicknesses (list(float)): list of the thicknesses of each layer. The top layer starts at x = 0.
-
densities (list(list(float))): number densities in
1/length_unit^3for each each species in[material_parameters]for each layer.
- length_unit (string): unit of length quantities in Mesh2D input section. Can be one of ANGSTROM, NM, MICRON, CM, MM, M, or an arbitrary float, e.g. "1e-6", for a value in meters
- energy_barrier_thickness: distance away from surface that particles interact with the surface binding energy. Recommended: ~n^(-1/3) This is in units [length_unit]
- points: (list(list(float, float))): (x, y) list of vertices that make up the mesh.
- boundary: (list(int)): indices of vertices that define the boundary of the mesh.
-
triangles: (list(list(int, int, int))): indices of the three vertices from
pointsthat make up each triangle. - densities (list(list(float)): list of number densities of each material species in each triangle. This is in units [atom/length_unit^3]
- simulation_boundary_points (list((float, float))): (x, y) for each point on the simulation boundary. Particles outside of the simulation boundary will be considered to have left the simulation and have been reflected or sputtered as appropriate.
- electronic_stopping_correction_factors (list(float)): correction factor for low-energy electronic stopping in the material. Recommended: 1.0 for each triangle unless known for a specific element/compound
The TriMesh geometry in RustBCA is a triangular mesh. 3D geometry is handled using the parry3d crate.
- length_unit (string): unit of length quantities in TriMesh input section. Can be one of ANGSTROM, NM, MICRON, CM, MM, M, or an arbitrary float, e.g. "1e-6", for a value in meters
- energy_barrier_thickness: distance away from surface that particles interact with the surface binding energy. Recommended: ~n^(-1/3) This is in units [length_unit]
- vertices: (list(list(float, float, float))): x, y, z for each vertex that makes up the mesh.
- indices: (list(list(int, int, int))): indices of vertex 1, vertex2, vertex3 for each triangle that makes up the mesh.
- densities (list(float)): list of number densities of each material species in each triangle. This is in units [atom/length_unit^3]
- electronic_stopping_correction_factors (list(float)): correction factor for low-energy electronic stopping in the material. Recommended: 1.0 for each triangle unless known for a specific element/compound

The TriMesh is a homogeneous composition, arbitrary geometry triangular mesh. TriMeshes are specified using a list of 3D vertex coordinates (vertices) and a list of indices of the vertices that make up each triangle. See the separateWiki page on the TriMesh feature for more information and examples.
The 2D homogeneous mesh is similar to Mesh2D, but has no internal inhomogeneous composition. Thus, it only has a list(float) for densities, a single electronic stopping correction factor, and only a list of points in points that define the polygonal shape that makes up the mesh. Currently, this option does not work with holes or self-intersecting polygons. Not only are these input files much easier to make, the homogeneous 2D mesh will be much faster for complex shapes.
HOMOGENEOUS2D is available on the dev branch; an example can be found here.
- length_unit (string): unit of length quantities in Mesh2D input section. Can be one of ANGSTROM, NM, MICRON, CM, MM, M, or an arbitrary float, e.g. "1e-6", for a value in meters
- points: (list(list(float, float))): (x, y) list of vertices that make up the target.
- densities (list(float)): list of number densities of each material species in the target. This is in units [atom/length_unit^3]
- simulation_boundary_points (list((float, float))): (x, y) for each point on the simulation boundary. Particles outside of the simulation boundary will be considered to have left the simulation and have been reflected or sputtered as appropriate.
- electronic_stopping_correction_factor (float): correction factor for low-energy electronic stopping in the material. Recommended: 1.0 for each triangle unless known for a specific element/compound
[1] W. Eckstein, Computer Simulation of Ion-Solid Interactions, p. 94-95 (1991)
[2] J.P. Biersack & L.G. Haggmark, A Monte Carlo Computer Program for the Transport of Energetic Ions in Amorphous Targets, Nuc. Inst. & Meth. 174 (1980) Eqs. 27-33
[3] J. Lindhard & Scharff, Energy Dissipation by Ions in the keV Region, Phys Rev., 124, 128 (1961)
[4] O.S. Oen & M.T. Robinson, Computer studies of the reflection of light ions from solids, Nucl. Inst. and Meth., 132 (1976)
[5] J. Lindhard & A. Winther, Stopping Power of Electron Gas and Equipartition Rule, Mat. Fys. Medd. Dan. Vid. Selsk. 34 4 (1964)
[6-9] W. Eckstein, Computer Simulation of Ion-Solid Interactions, p. 40-62 (1991)
[10] M.H. Mendenhall, R.A. Weller, Algorithms for the rapid computation of classical cross sections for screened Coulomb collisions, Nucl. Inst. Meth. Phys (1991)
[11] J.F. Ziegler, J.P. Biersack, and M.D. Ziegler, SRIM (2012)