{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
-01-GScr.png){kind=link}
-01.png){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
This website gives the possibility to use OpenCalphad using a virtual machine. In computing, a virtual machine (VM) is an emulation of a computer system. The virtual machine of OpenCalphad was designed to run on the platforms: Windows, Mac, and Linux. It uses the host software of the VirtualBox project, which is has the GNU Licensing.
Screenshot #1: 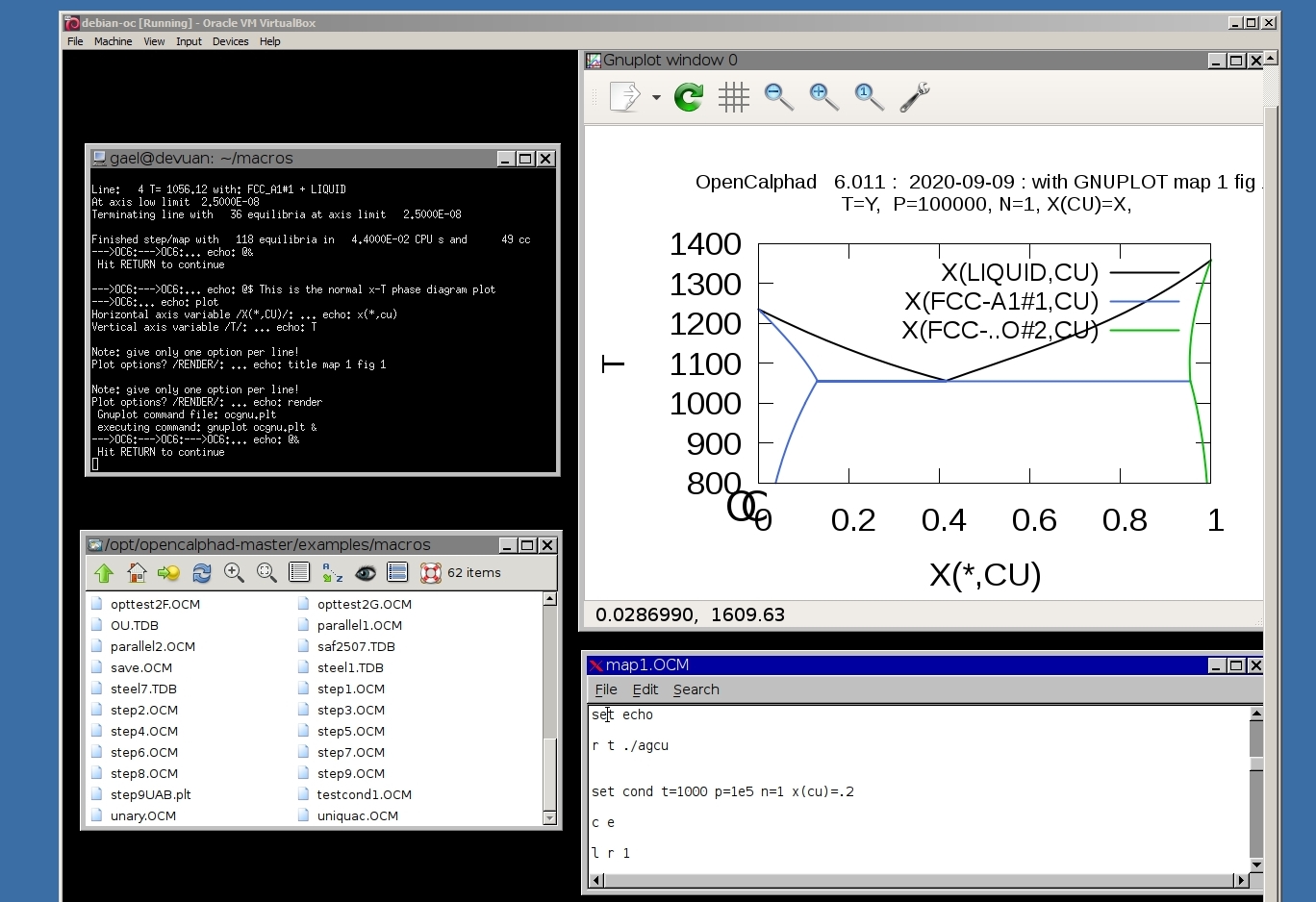
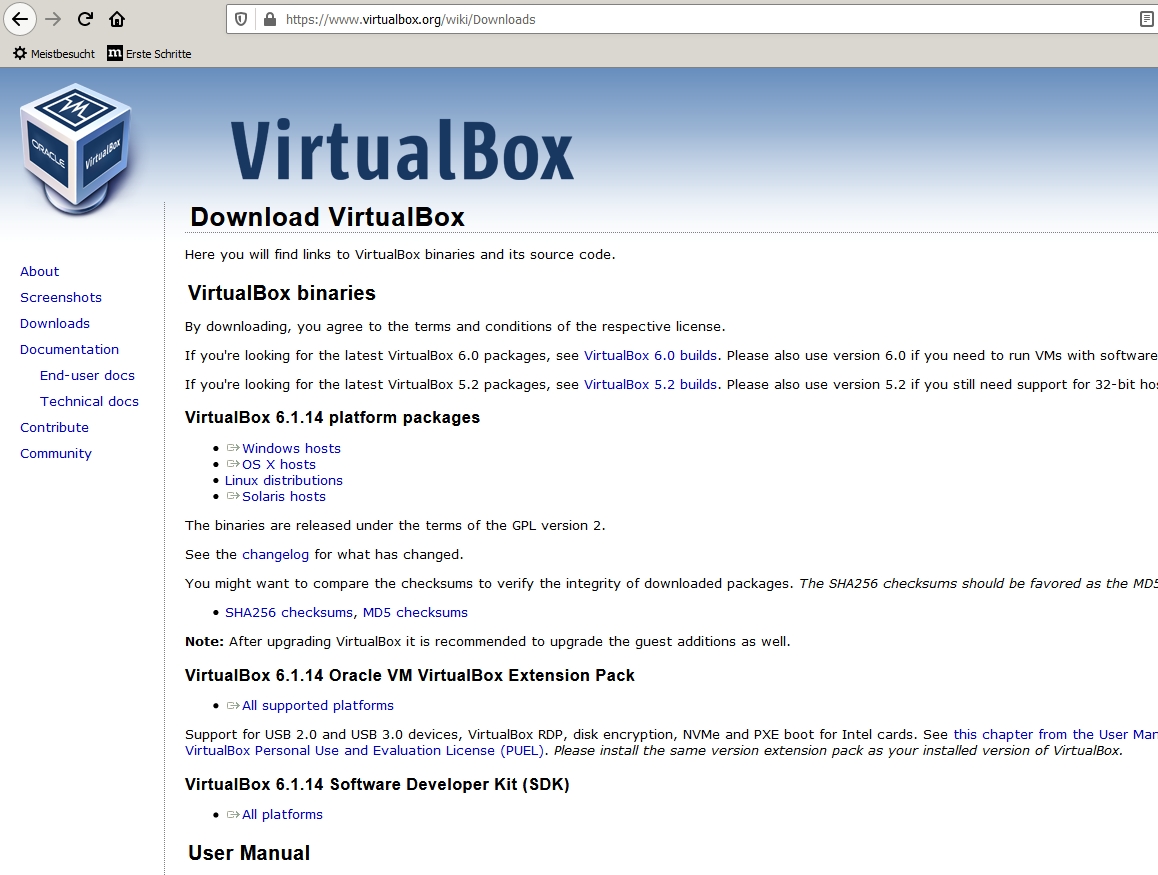
Download the software virtualbox corresponding to your architecture. https://www.virtualbox.org/wiki/Downloads If necessary, the manual of installation of virtualbox is at the following url: https://www.virtualbox.org/wiki/Documentation After download, virtualbox needs to be installed.
Screenshot #2:
Alternatively, the software can run as well under VMWARE VMPLAYER 16.
After the installation of virtualbox, you may find the image at the following link. URL to download the version of OpenCalphad for classroom: https://www.dropbox.com/s/4lab0ipan8e0rrw/virtualbox-debian-opencalphad.zip?dl=0
The image is destined to the console/terminal usage. The GUI applications are not provided in this above image. (See below for images with the OC with GUI).
After the download of the image file of the virtual machine, the files need to be uncompressed. The image is compressed with the regular ZIP compression format. It can be uncompressed with software, like Winzip or Winrar. The "debian-oc.vdi" file is the main file, which will be opened using virtualbox. This will result in the following screenshot (screenshot #3).
Screenshot #3:
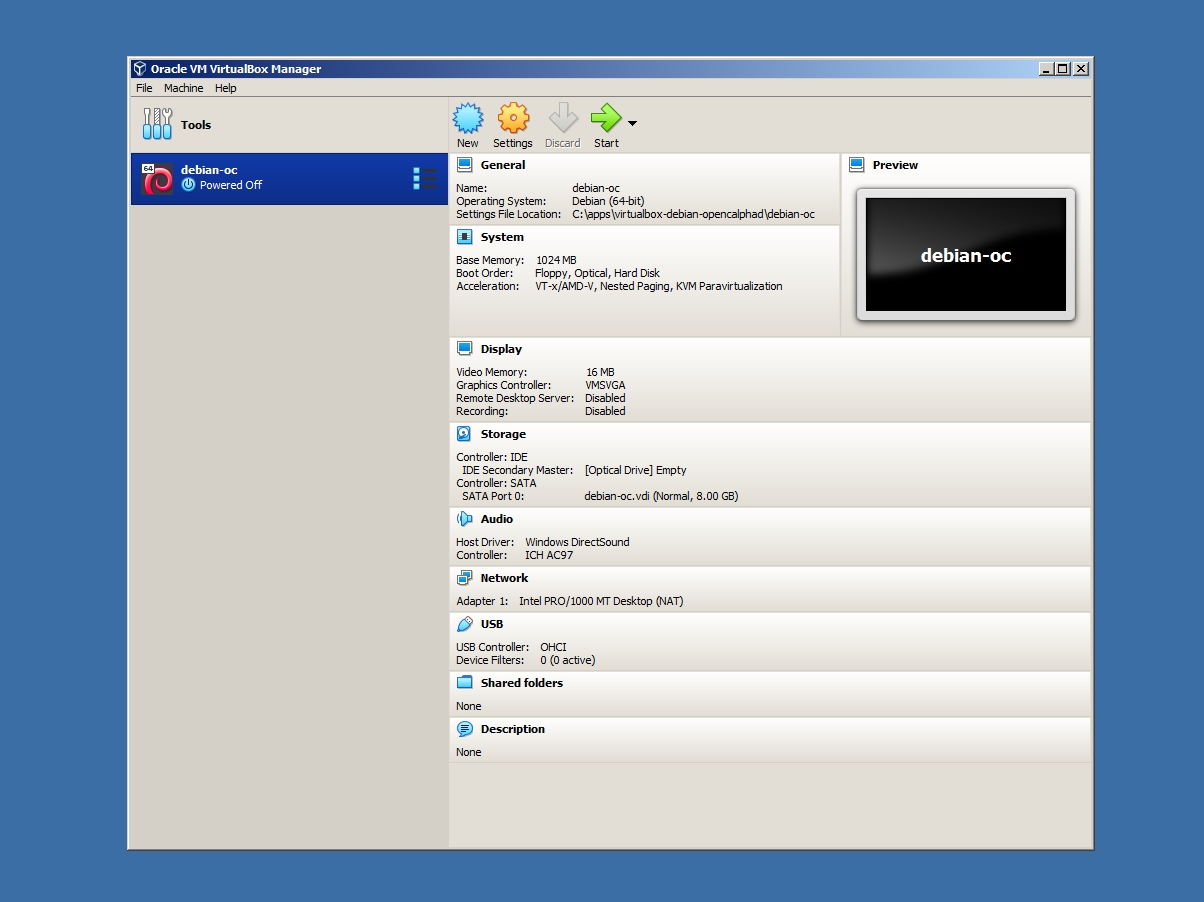
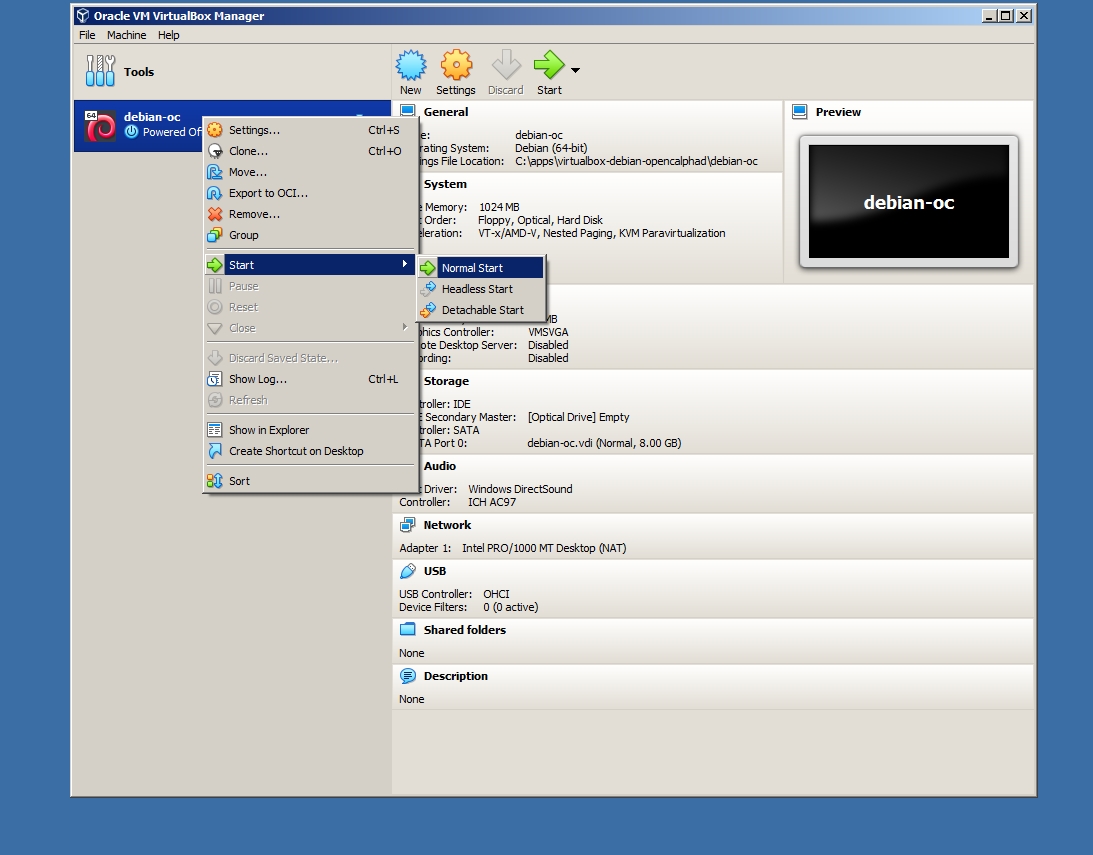
After the user can use the context menu of the virtual machine, as indicated in the screenshot (screenshot #4), to start the machine. "Start" will be displayed (screenshot #4). The virtual machine will boot. It will take 1-2 minute(s) of boot time. After the boot of the system, OpenCalphad will be launched directly. The virtual machine is running OC on the Linux operating system. The window manager is IceWM.
Screenshot #4:
The documentation of OpenCalphad is available at the url: http://www.opencalphad.com/
Please visit the website of OpenCalphad for more information.
In the terminal xterm, the command "oc" is available to load and run a macro.
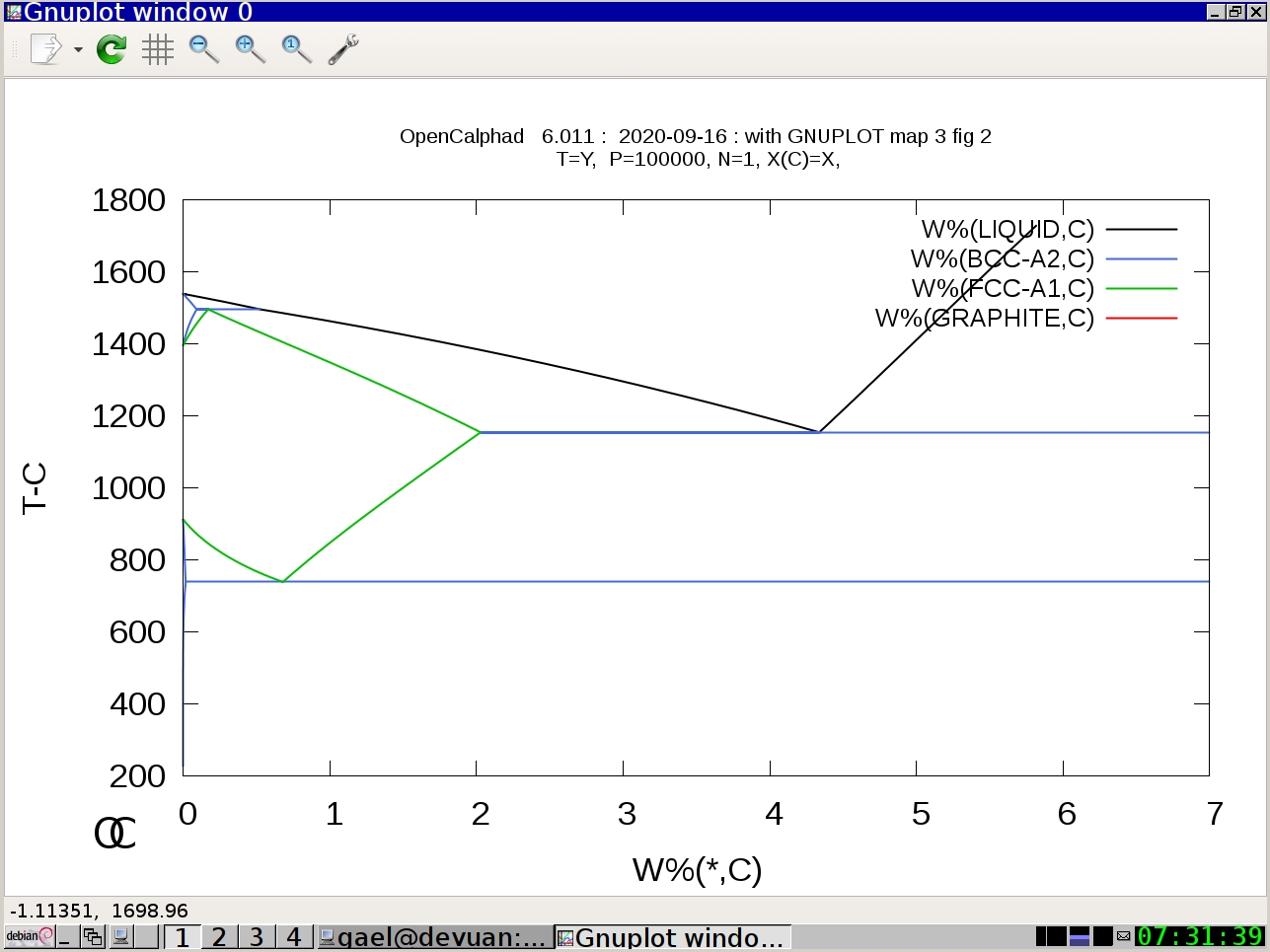
Example #1: Fe-C system
oc macros/map3.OCM
(Press Return)
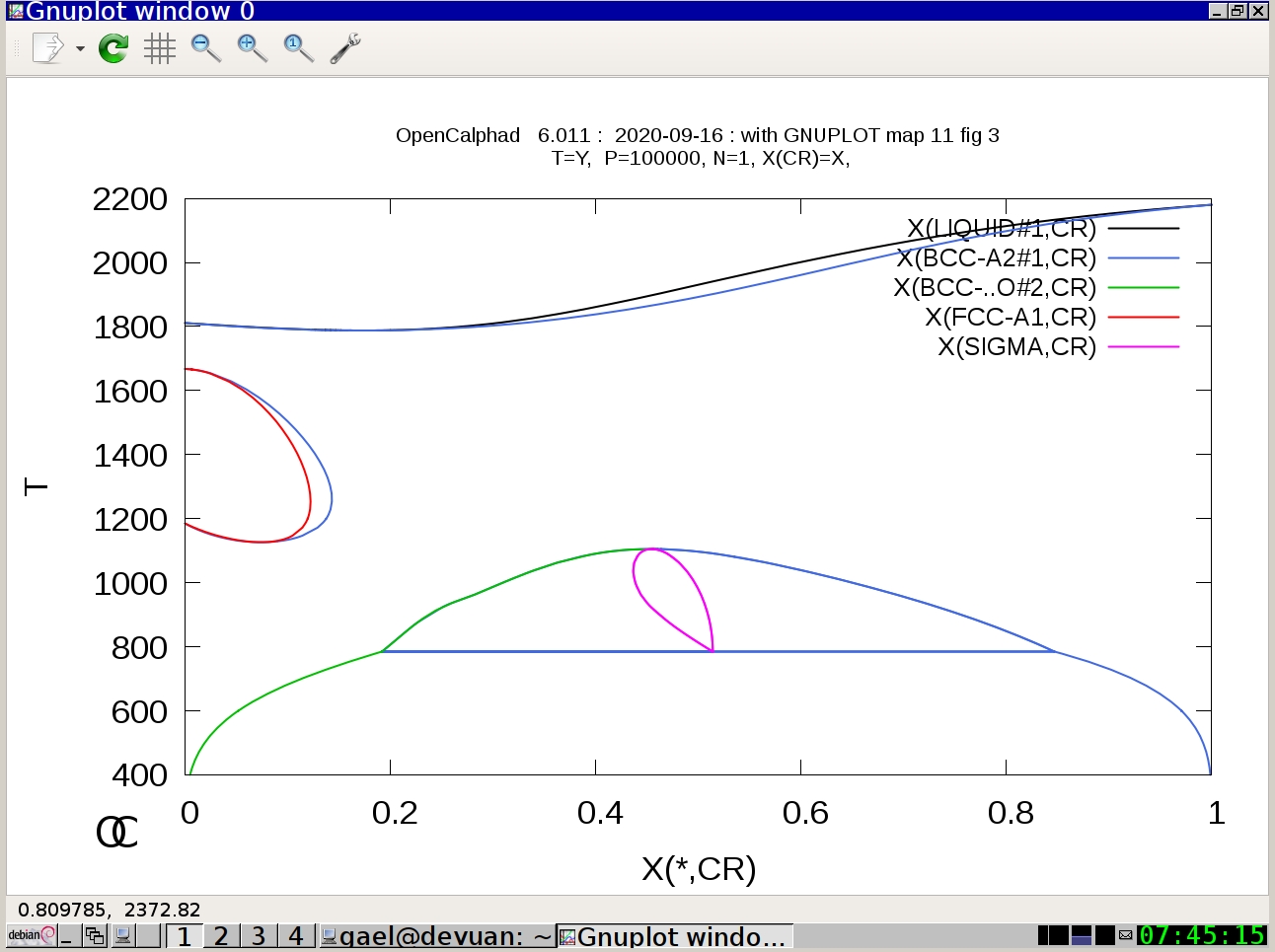
Example #2:
oc macros/map11.OCM
(Press Return)
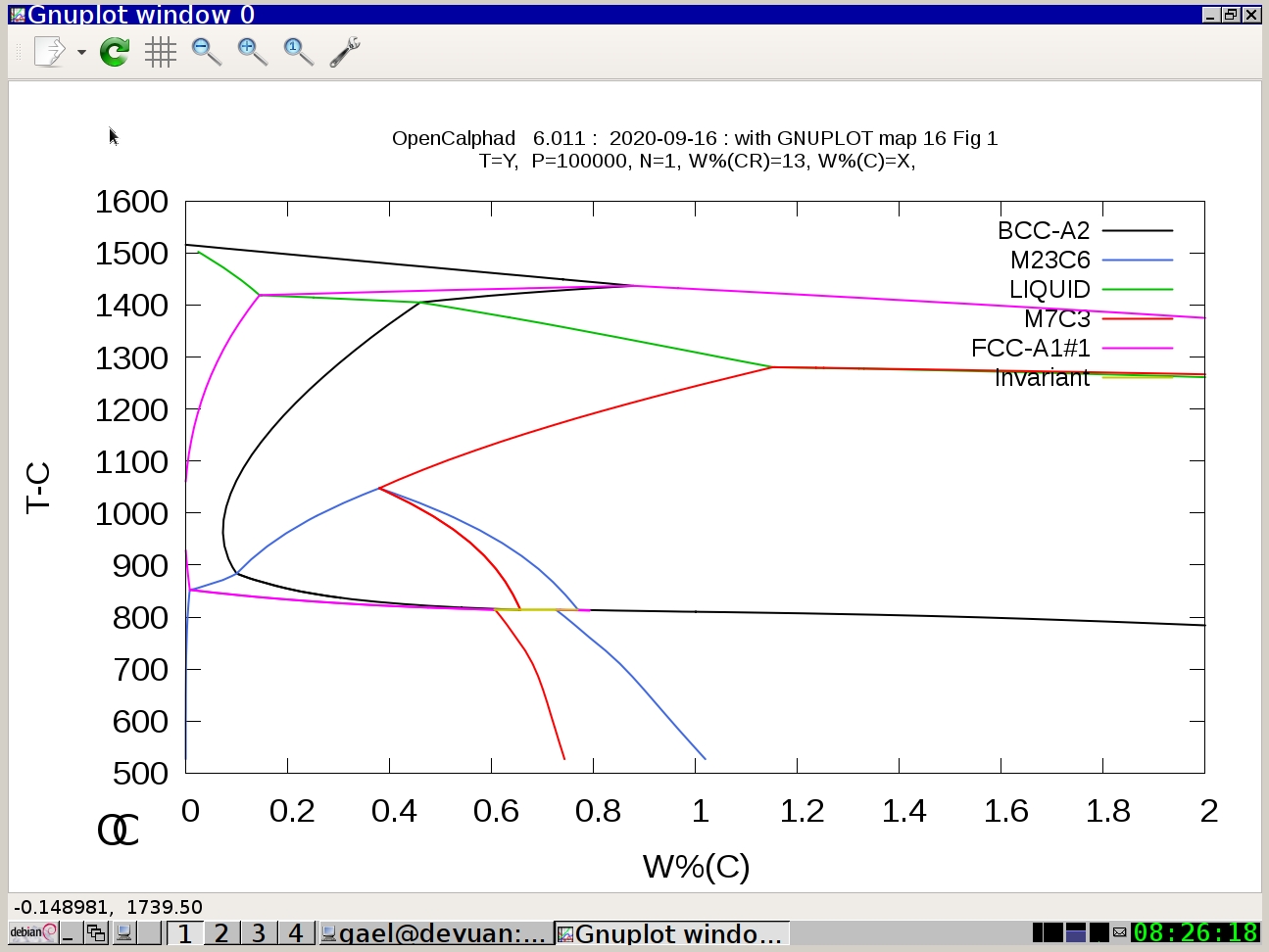
Example #3: Fe-C-Cr system
oc macros/map16.OCM
(Press Return)
Example #4: Al-Fe system
oc macros/map17.OCM
(Press Return)
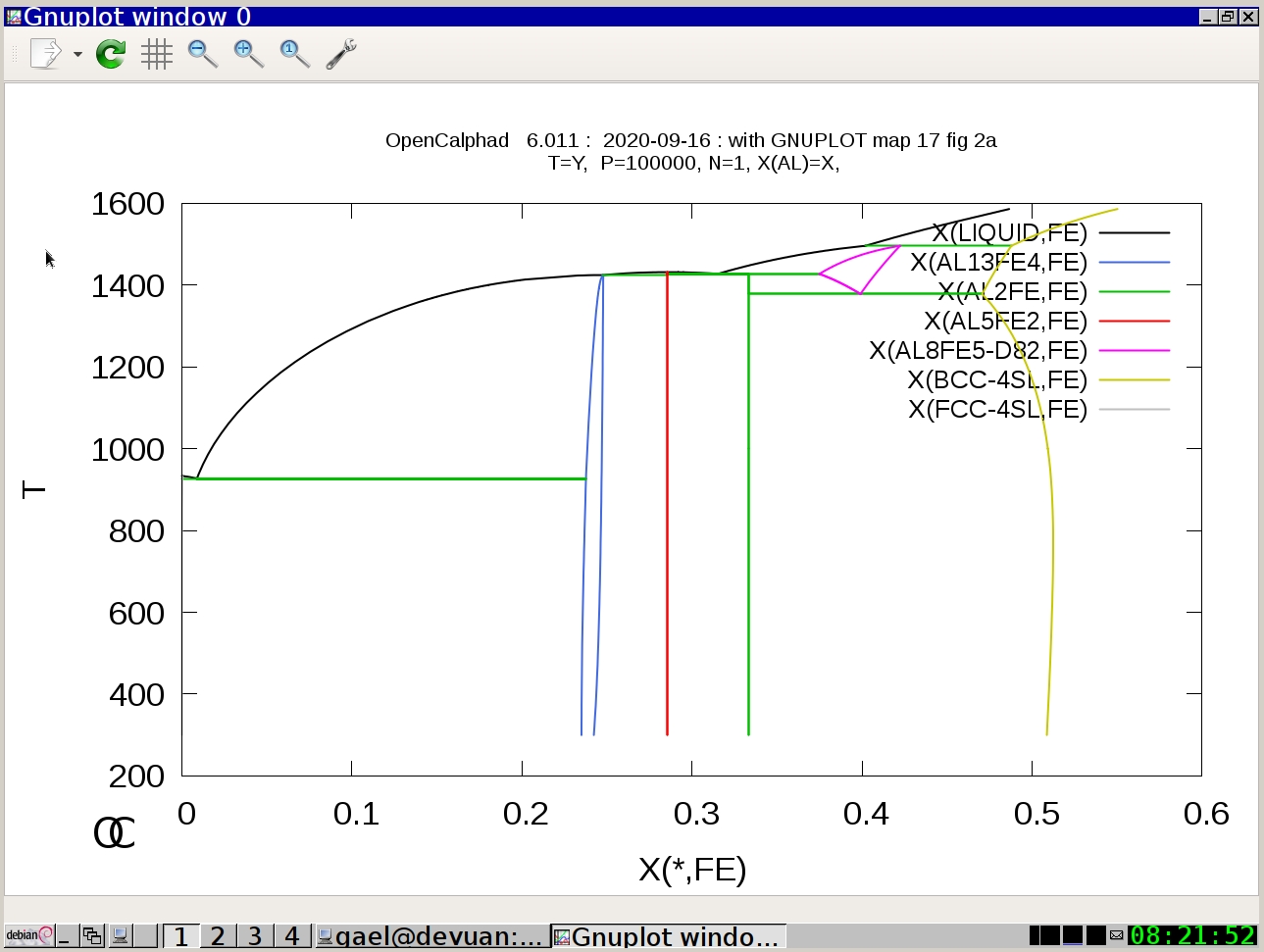
The graphical user interface (GUI) of OC is under development.
An image of the virtual machine with the GUI OC is available for download at the following Link.
The image of first release v0.1 is available at the following link: https://drive.google.com/file/d/14aJRksYz4pKwuMBolMNSc3sxOllR8QNZ/view?usp=sharing
(md5 checksum: 57ff8cc52de02868de4d255a9c6b140b virtualbox-debian-opencalphad-GUI-v1.1.zip)
The image of the second release v0.2 is available at the following link: https://drive.google.com/file/d/1QpcMUm6gPCDYn_kI7C0EAm74955F71OJ/view?usp=sharing
(md5 checksum: 453a7bd35393a1ef615c903c7526a20c virtualbox-debian-opencalphad-GUI-release-0.2.zip)
The image of the release v0.31 is available at the following link: https://drive.google.com/file/d/1CBgLcKOFzHgVwEf4Nt2ra_2HgwBKXZ9G/view?usp=sharing
(md5 checksum: 1780fa953a0099ea59770c7a31616bf4 virtualbox-debian-opencalphad-GUI-release-0.31.zip)
Release v0.32, 2021/06/06:
https://gitlab.com/lusamek/opencalphad/-/raw/main/version-3.2/virtualbox-debian-opencalphad-GUI-release-0.32.zip
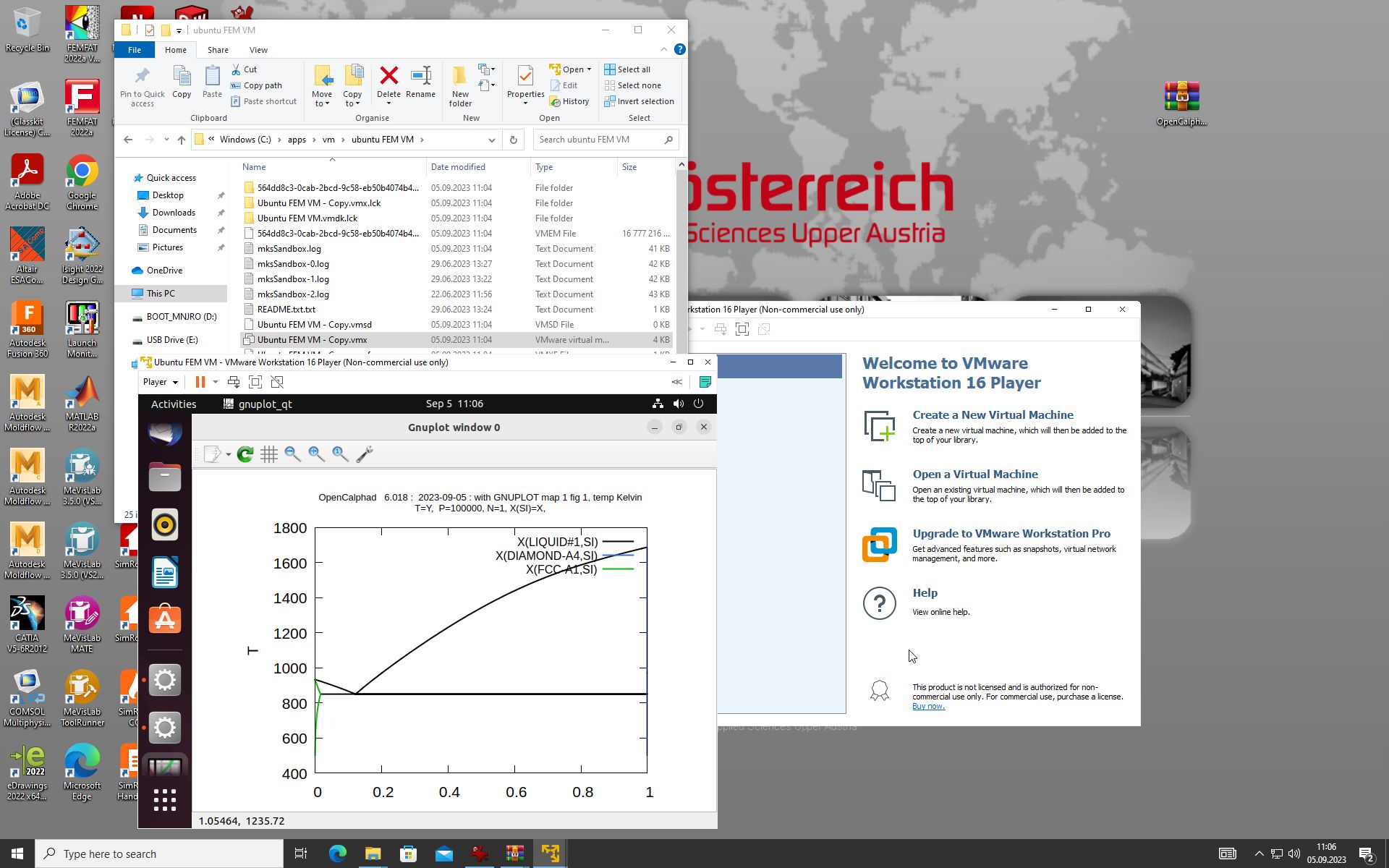
A VMDK image is as well available for use with QEmu (opensource) or Vmplayer (VMWare, prop. software).
Software: VMPLAYER (Vmware)
VMDK Datei (harddisk): https://gitlab.com/lusamek/opencalphad/-/raw/main/version-3.2/debian-oc-v0.32.vmdk
See more files and examples: https://gitlab.com/lusamek/opencalphad/
Virtualbox on Linux:
Kernel and headers for Ascii/Stretch: https://www.dropbox.com/sh/p21btpcbt8d0lf6/AABMbmwVGKGjm1LBvjPxgx7Ca?dl=0
Currently, the GUI can be compiled using the GNU C/C++ Compiler.
The "Makefile" allows to compile flopencalphad on Mac, Linux, and BSD/Unix.
This project is downloaded on the host machine or Unix/Linux/Mac.
The ZIP file needs to be decompressed. It requires the FLTK library and C++ compiler (e.g. CLANG or GCC).
In order to create the executables, the compilation is possible with make all.
The installation is possible with make install using root access. This will install the files into the directory /usr/local/bin/. The command echo $PATH will display the default PATH for executables. As user, the GUI can be started by the following command from the directory, where it was compiled :
./flopencalphad ~/macros
Screenshot of Virtual Machine running the GUI:
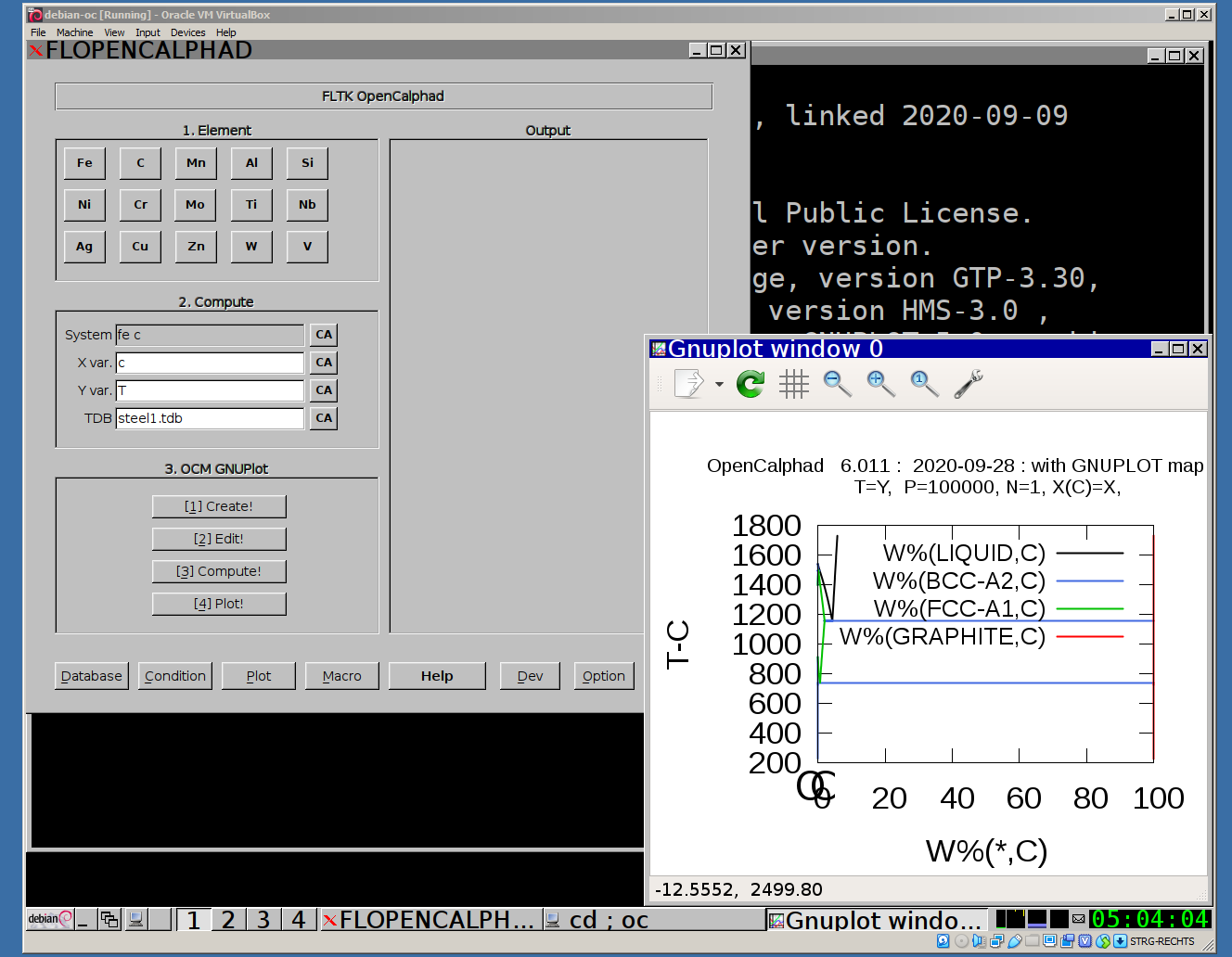
A live image for USB-Stick/Pendrive with OC and some utils for chemistry / material sciences is available.
The image can be copied on a pendrive or directly on harddisk. It contains the distribution OpenSUSE and OpenCalphad. The utility allows to calculate and compare results with the web. It is bootable on PC using a pendrive with the EFI / UEFI.
It requires an USB-Stick/pendrive larger than 32 GB (>= 32GB). To create the live pendrive: zcat image-opensuse-leap-master-amd64-axel-OC-kalzium-version1.1.img.gz > /dev/sdX
On Windows, you may use rawrite. https://www.netbsd.org/~martin/rawrite32/download.html
It is beta / early testing and with few features. It allows to use OC, Kalzium for chemistry,...

- Installation running on VMPlayer VMWARE
Download link for the client (windows): https://download3.vmware.com/software/WKST-PLAYER-1625/VMware-player-full-16.2.5-20904516.exe

(Download link will be available)
flopencalphad ~/macros/
(Press Return)
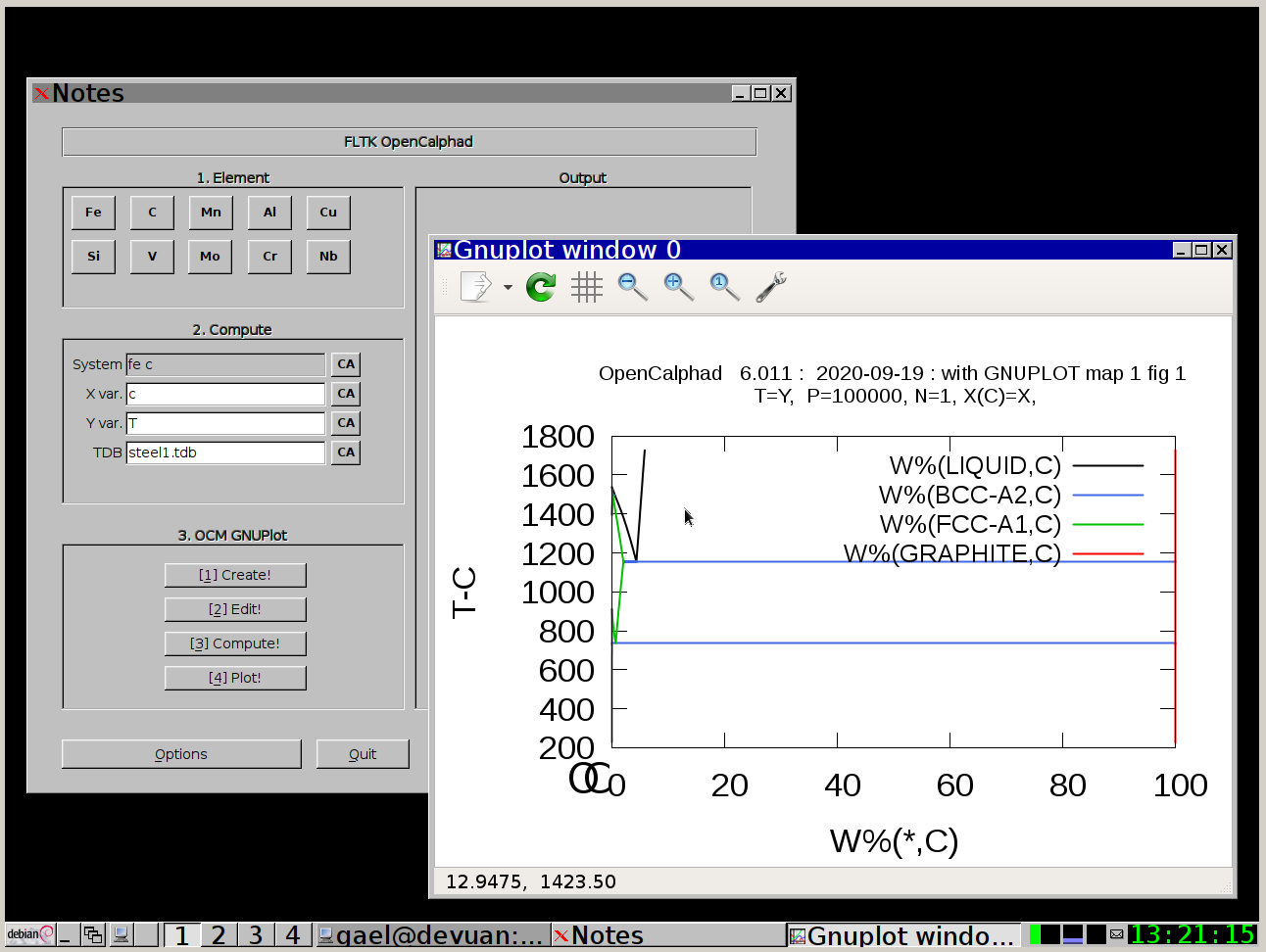
Frontend/GUI:
flopencalphad ~/macros/
(Click on "Macro" button)
The "Database" panel allows to select the database (*.tdb file). It allows to list the elements and species, which are available in the database.
The "Macro" panel gives a preview of the list of commands.
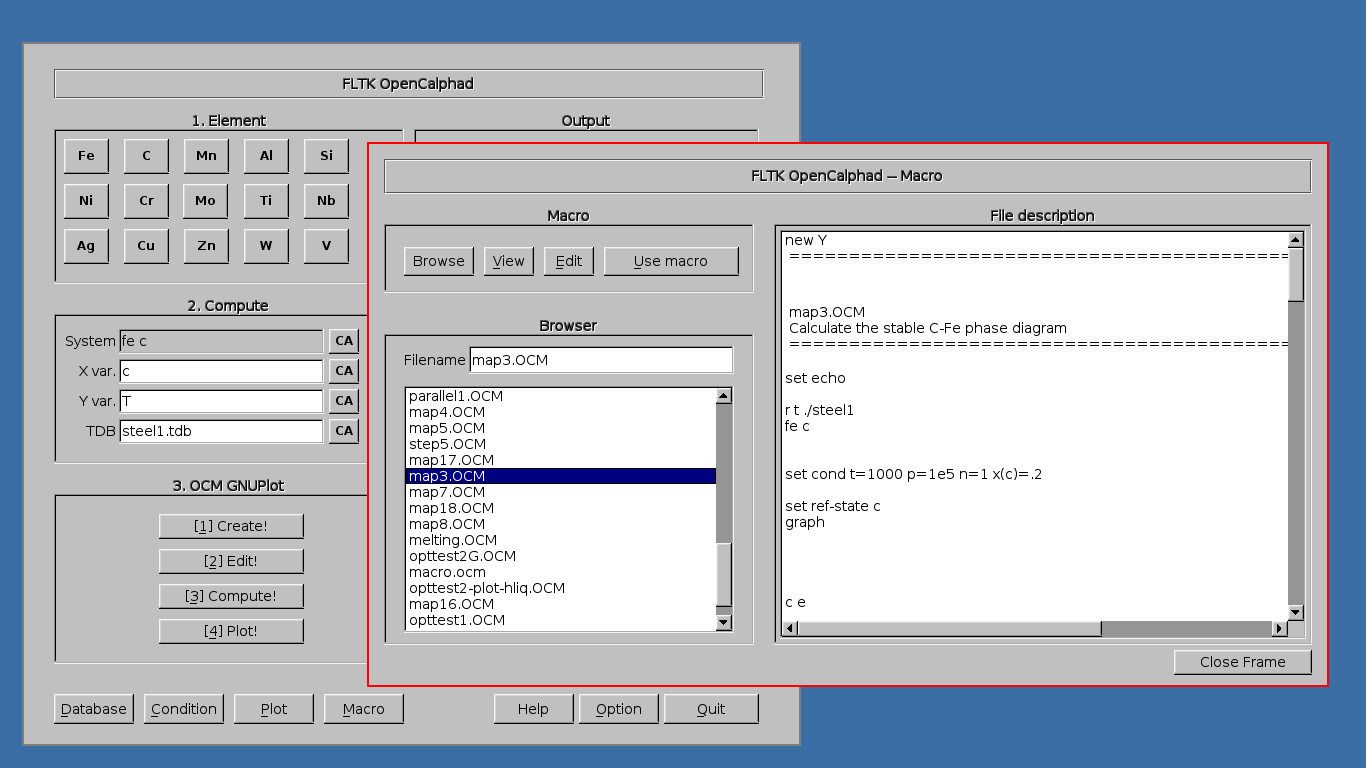
The "Macro" panel allows to edit and run a list of commands. The button "use macro" will select and setup the macro for the simulations. After selection of the macro, the process "2. Edit", "3. Compute", and "4. Plot" will display the results of the simulations.
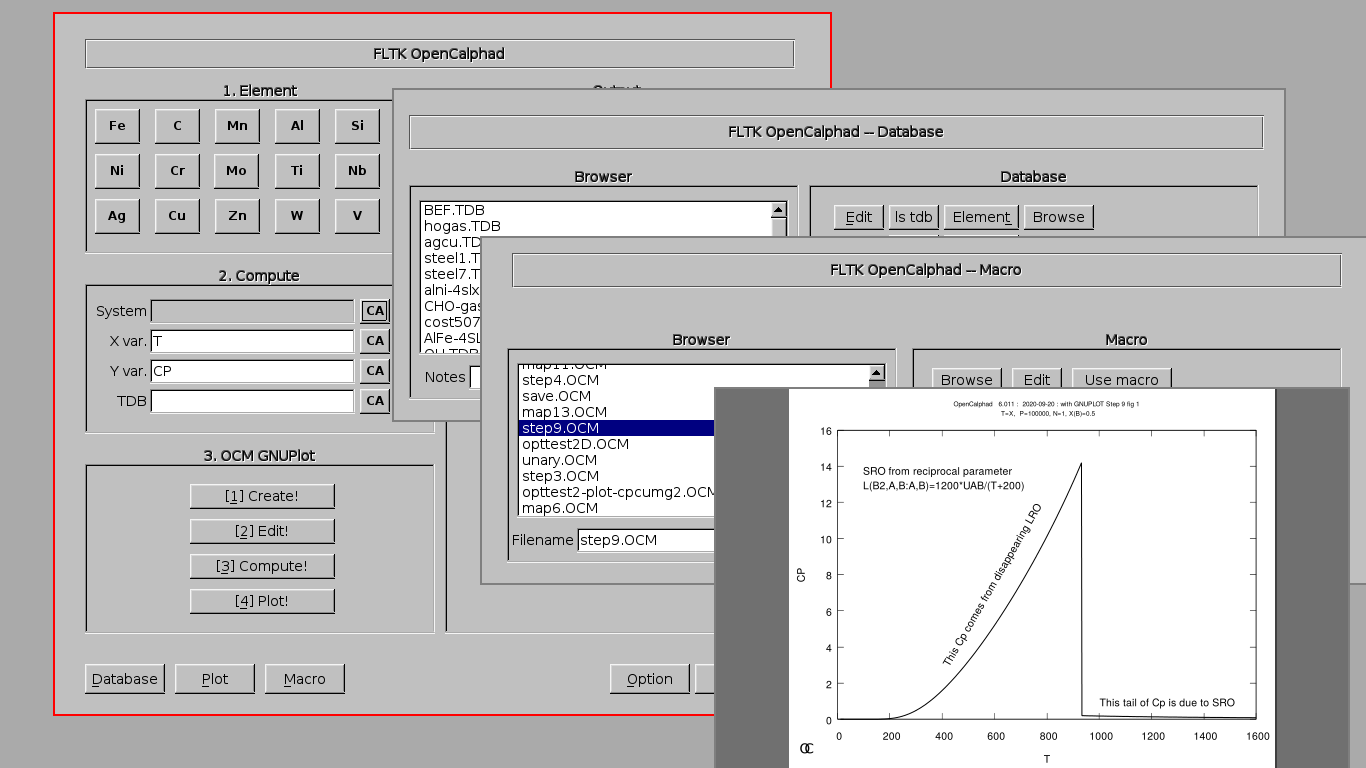
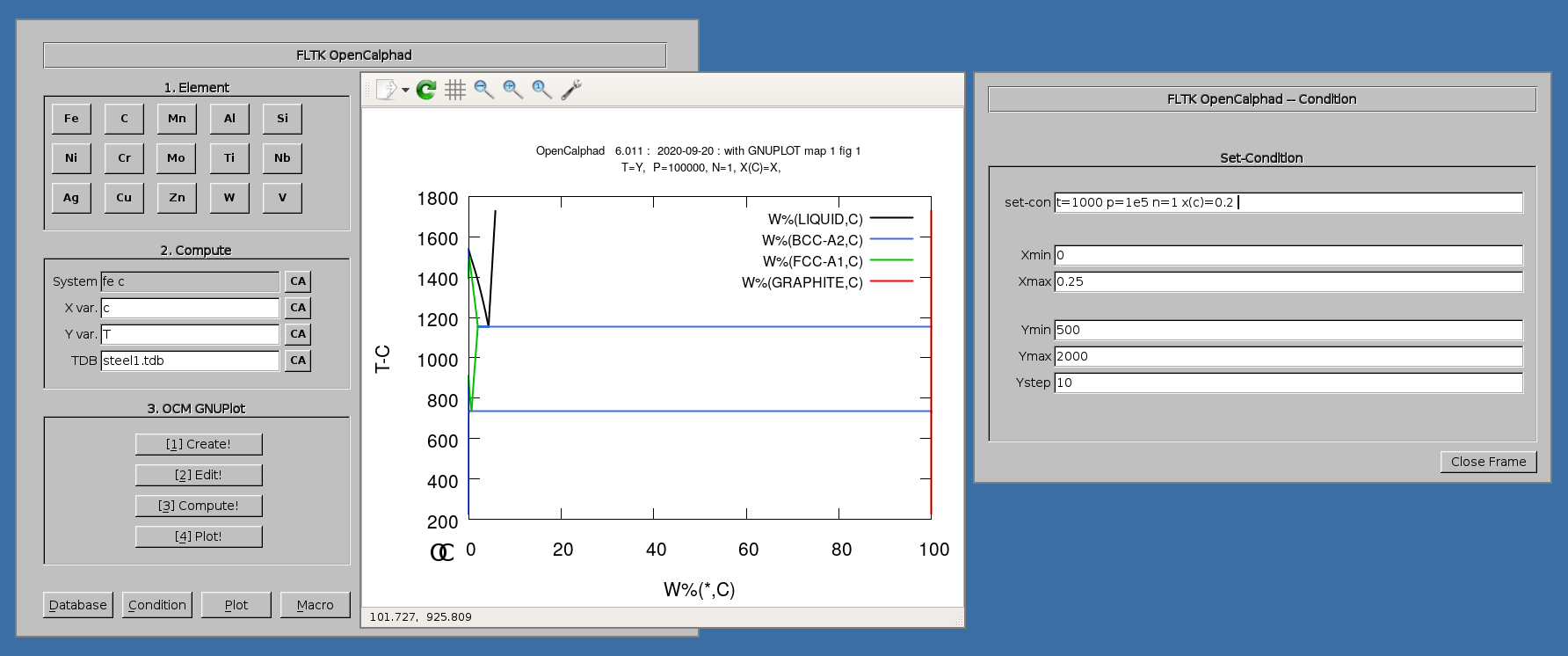
The panel for "Set-Condition" allows to give the initial parameters.
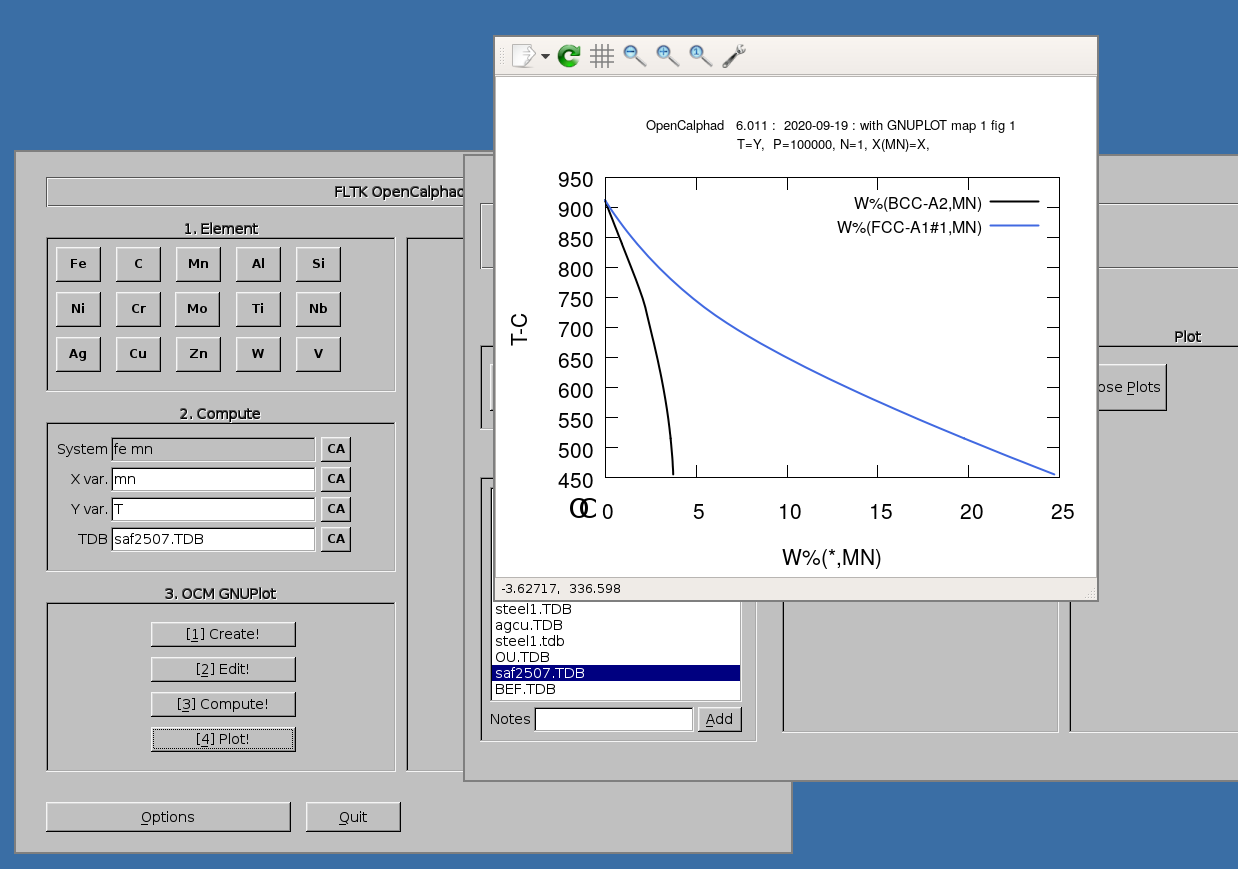
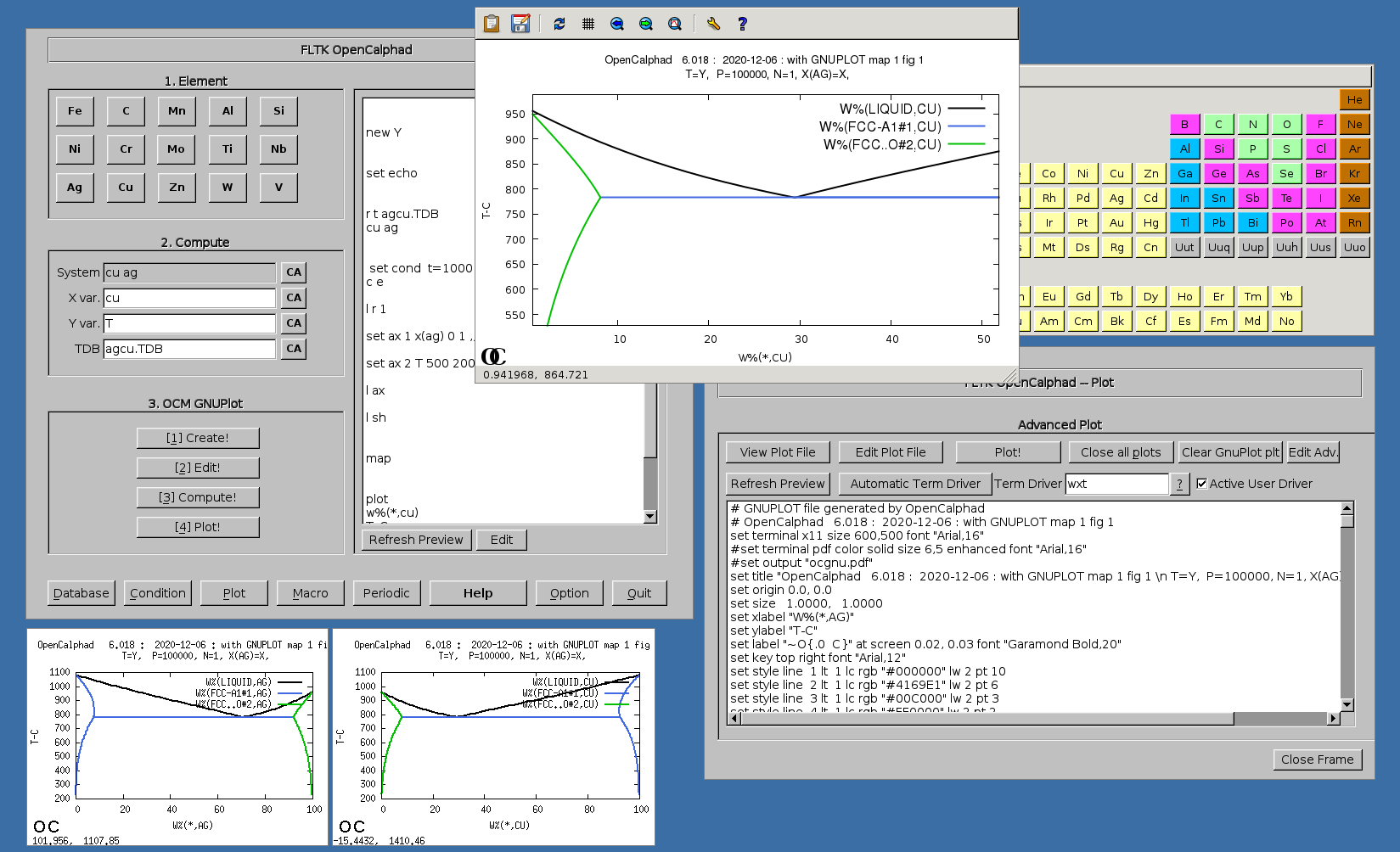
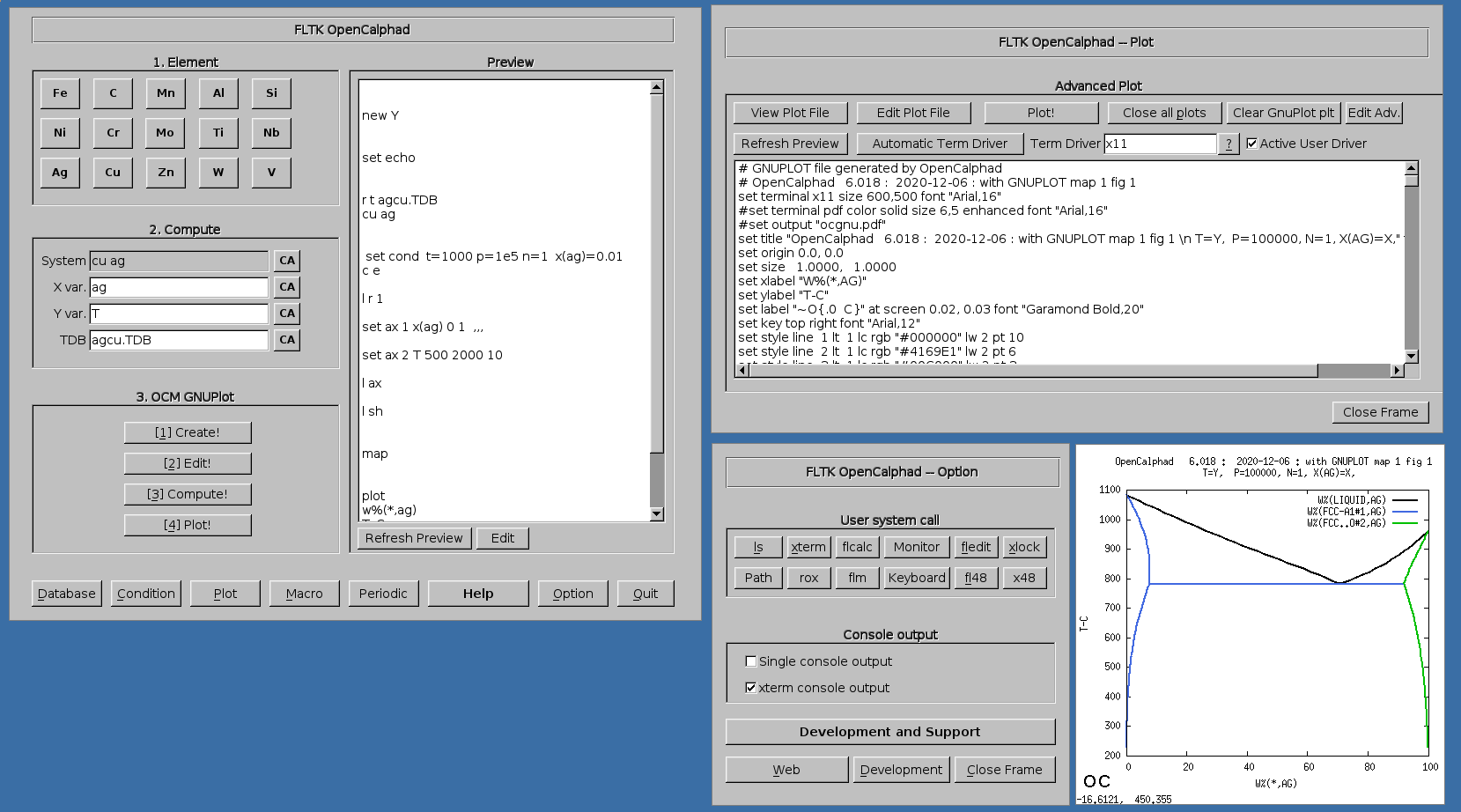
The GUI allows to use gnuplot-x11 (or gnuplot-qt) to plot the results. The results can be viewed and there is a zoom option with gnuplot-x11. This allows to use use the cursor for the position (x,y) as well. An example is shown for the Ag-Cu system:
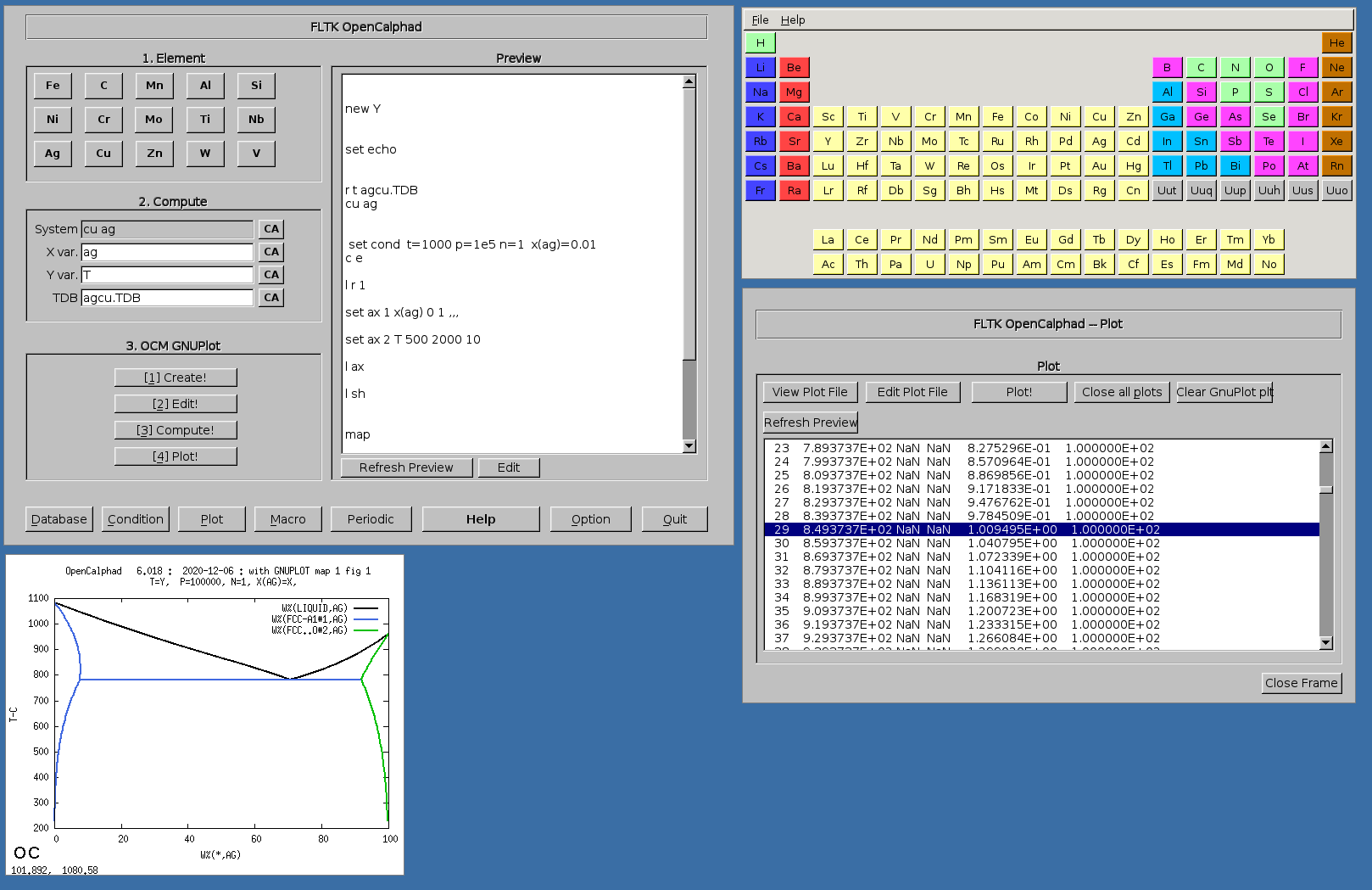
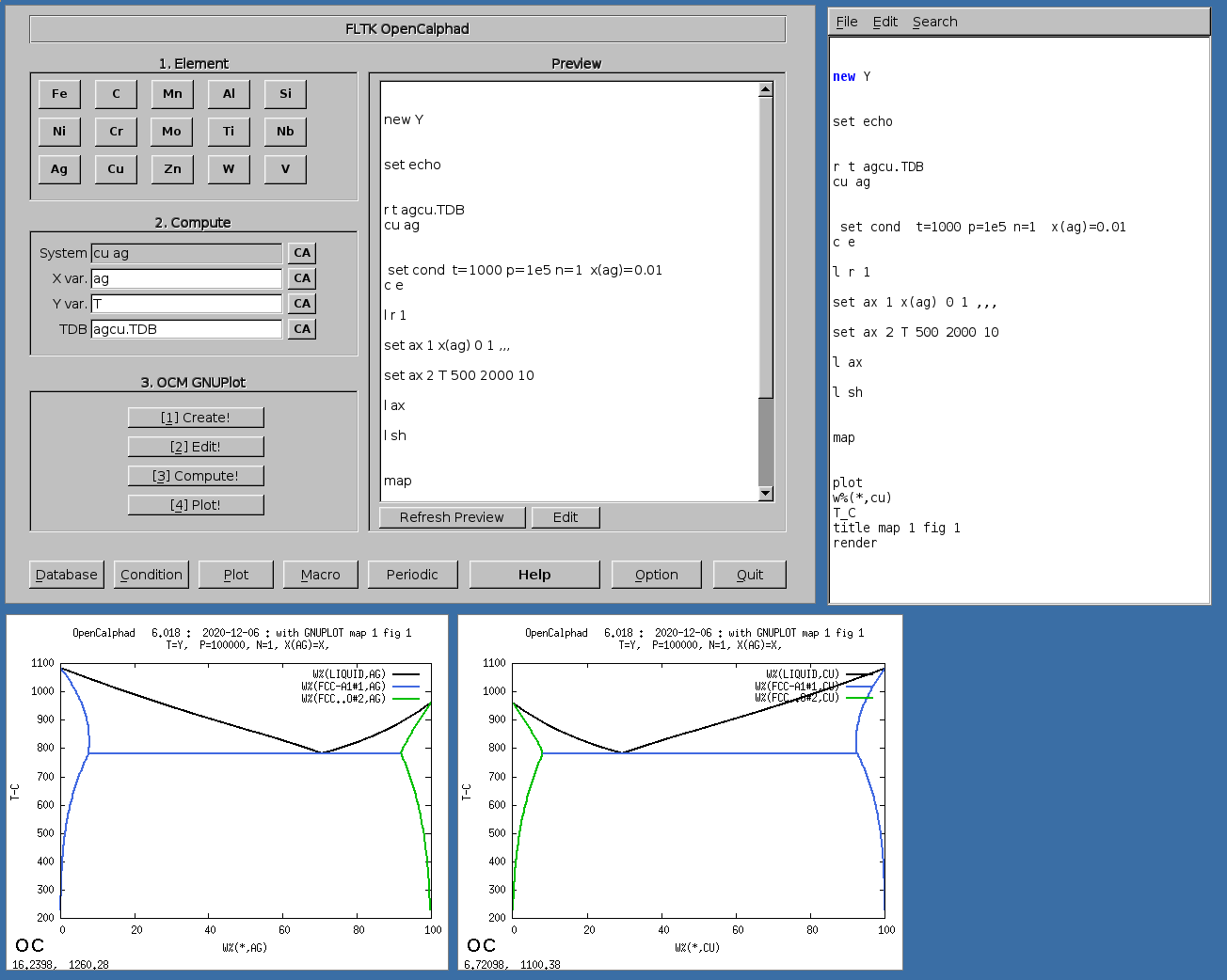
The Fe-C phase diagram can be calculated with OC.
The possibility to overlay different diagrams is as well given.
Plot the metastable Fe-C diagram and overlay the stable:
Fe-C phase diagram (metastable):
-01.png)
Fe-C phase diagram (metastable) with GUI:
-01-GScr.png)
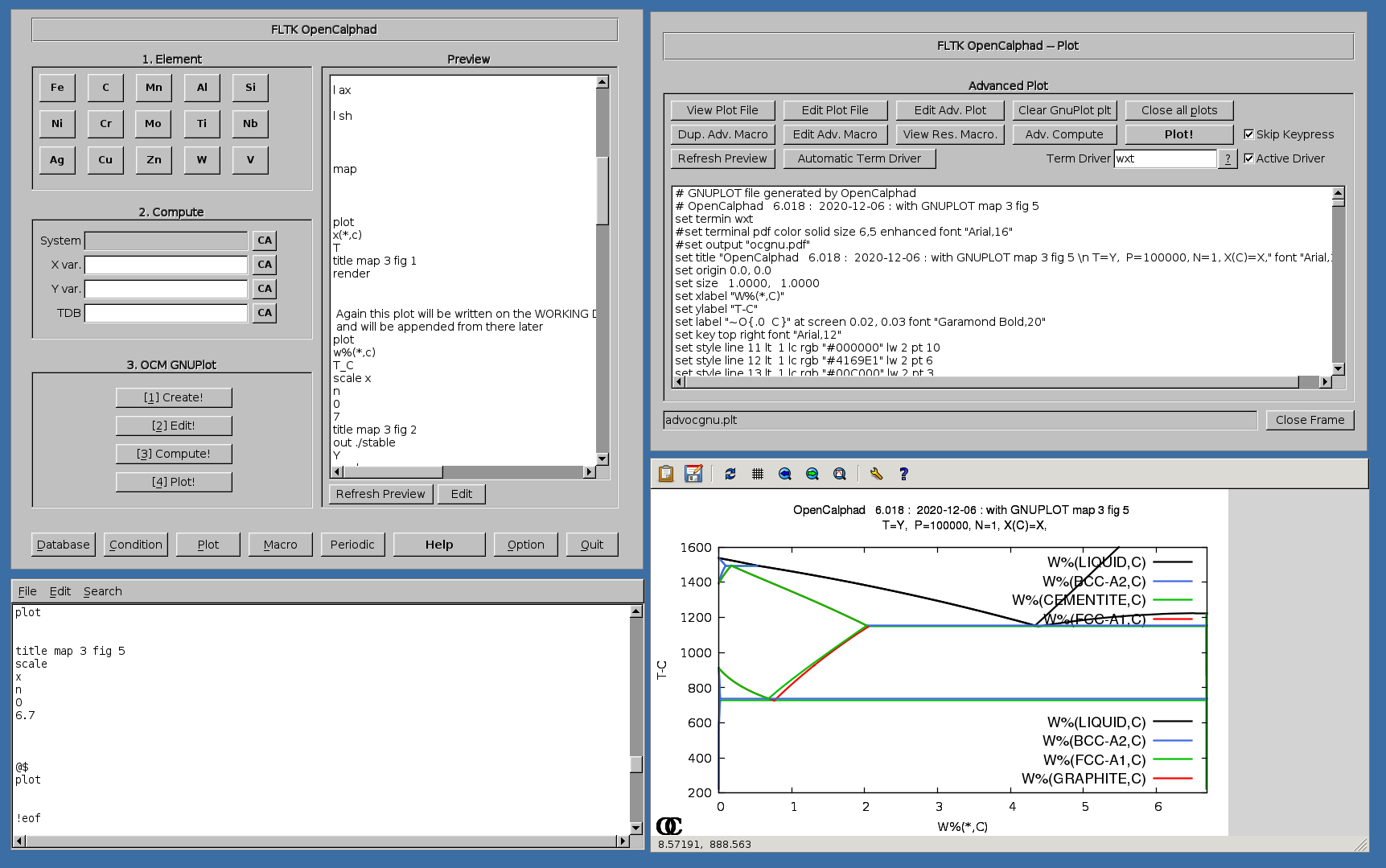
Phase diagram can be studied using OC.
Example of calculations:

The plot can be changed in order to study the phase diagram. The user select a given X/Y scale and click on "Plot".
Some examples for Fe-C, Cu-Ni, and Ag-Cu systems are shown in following Figures.
The macro for Ag-Cu is provided in this website under the directory "macros".
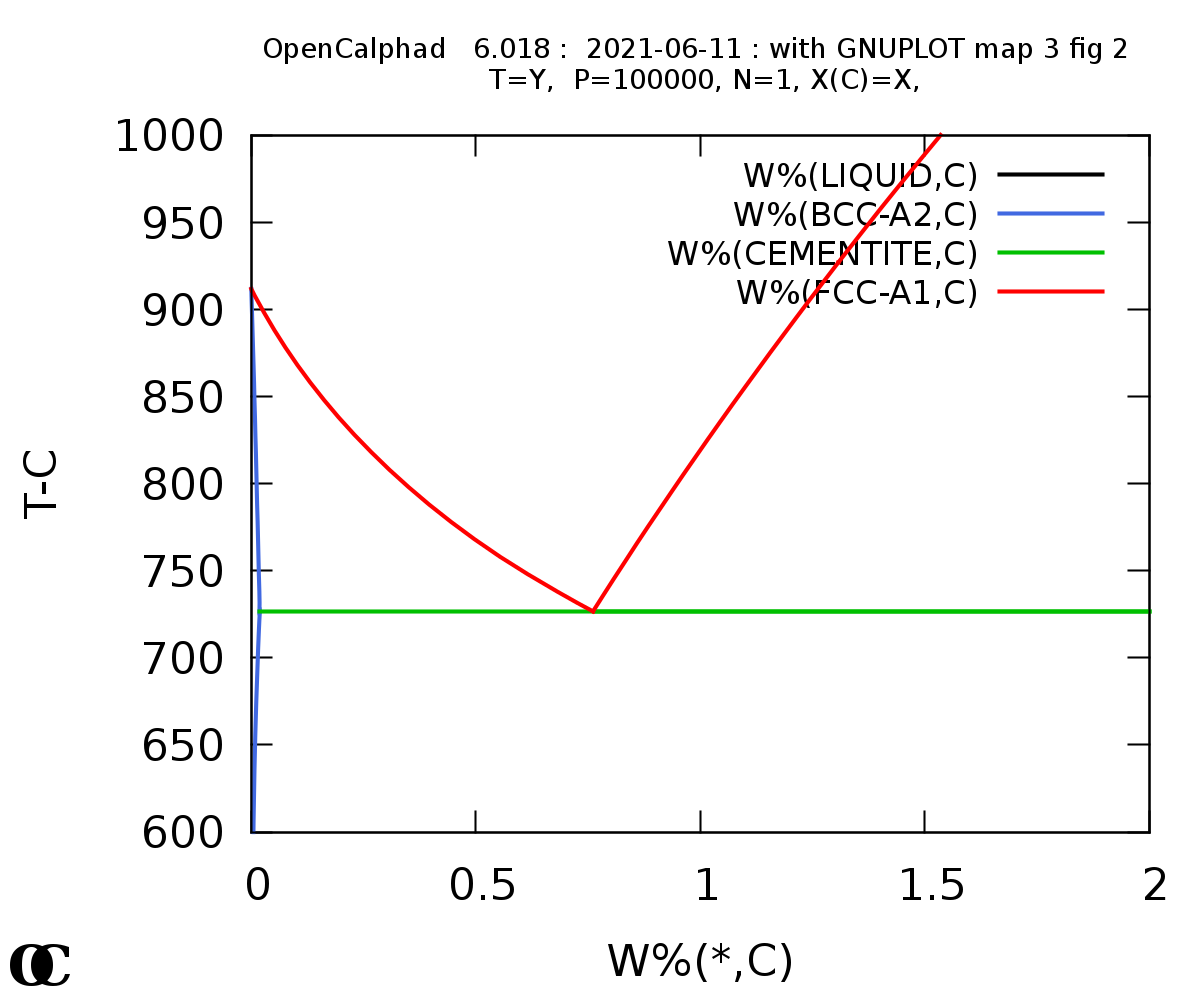
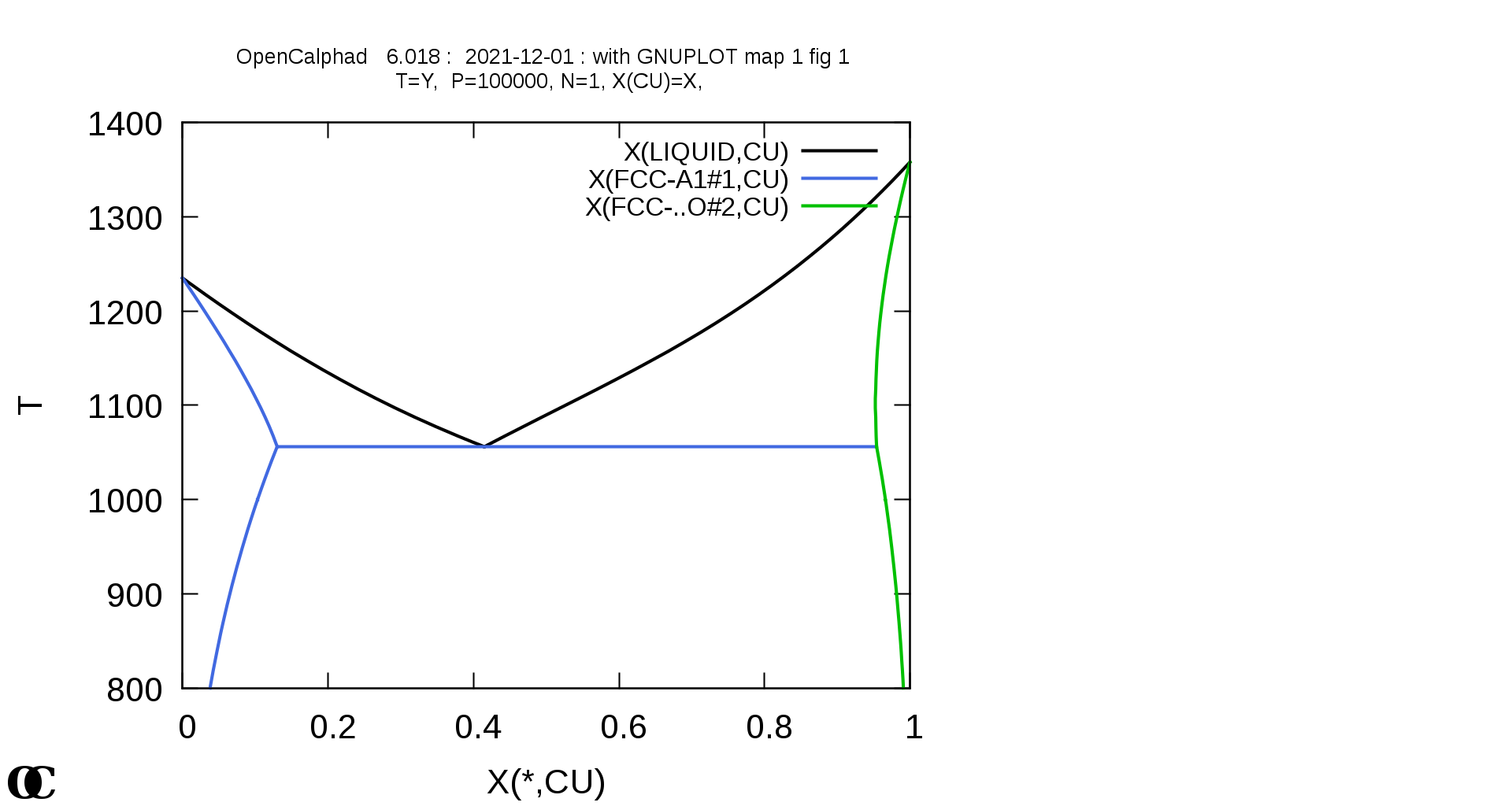
The macro for Ag-Cu is provided in this website under the directory "macros".
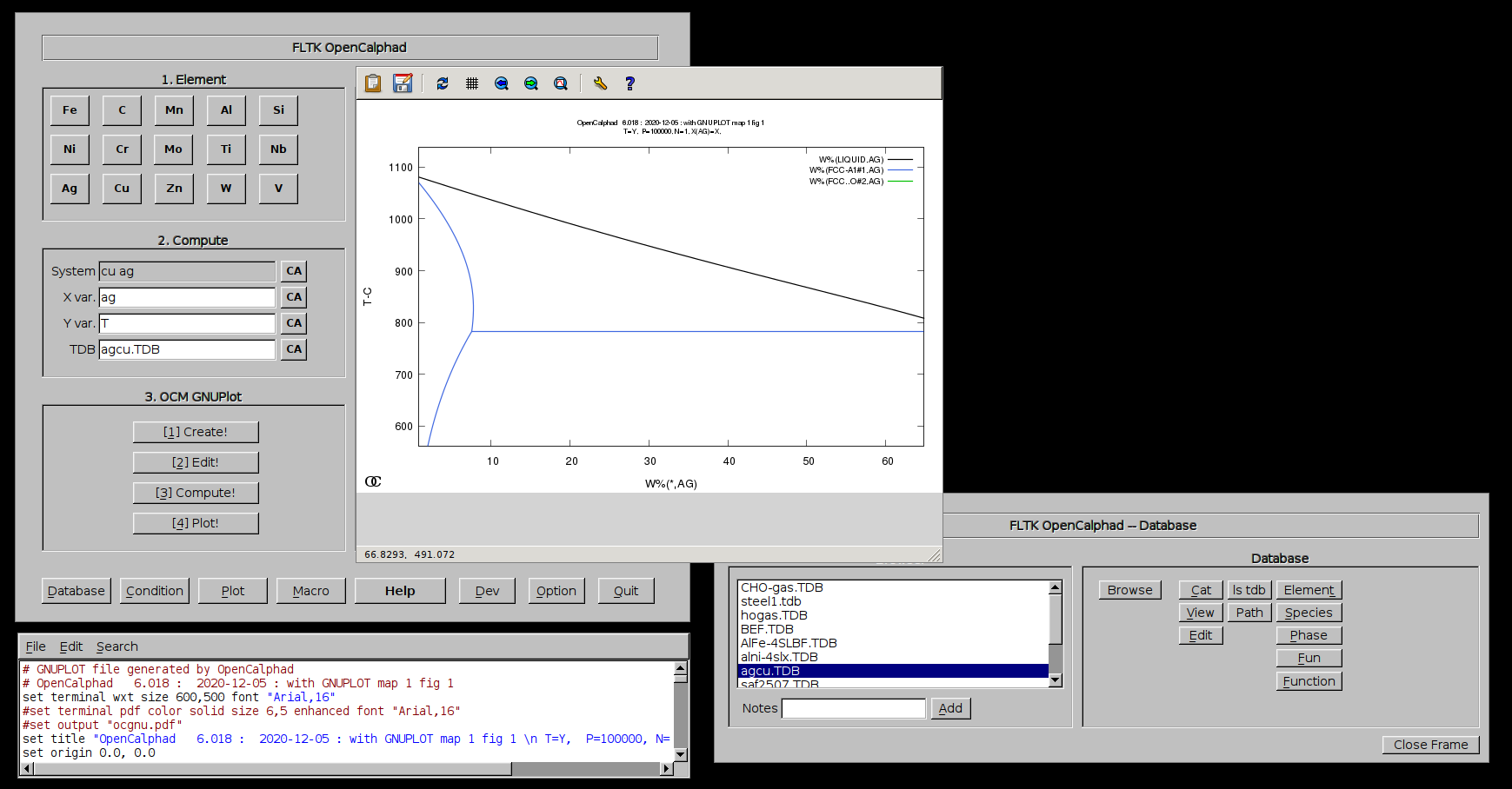
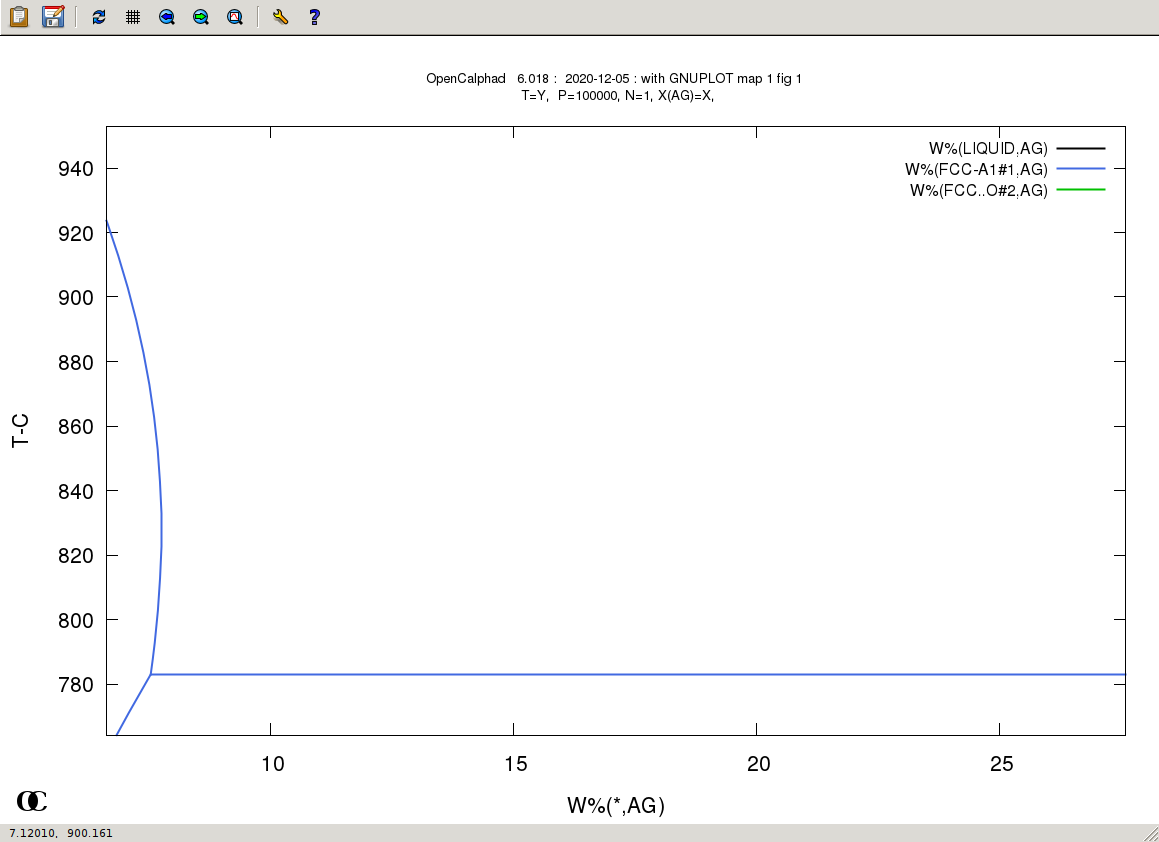
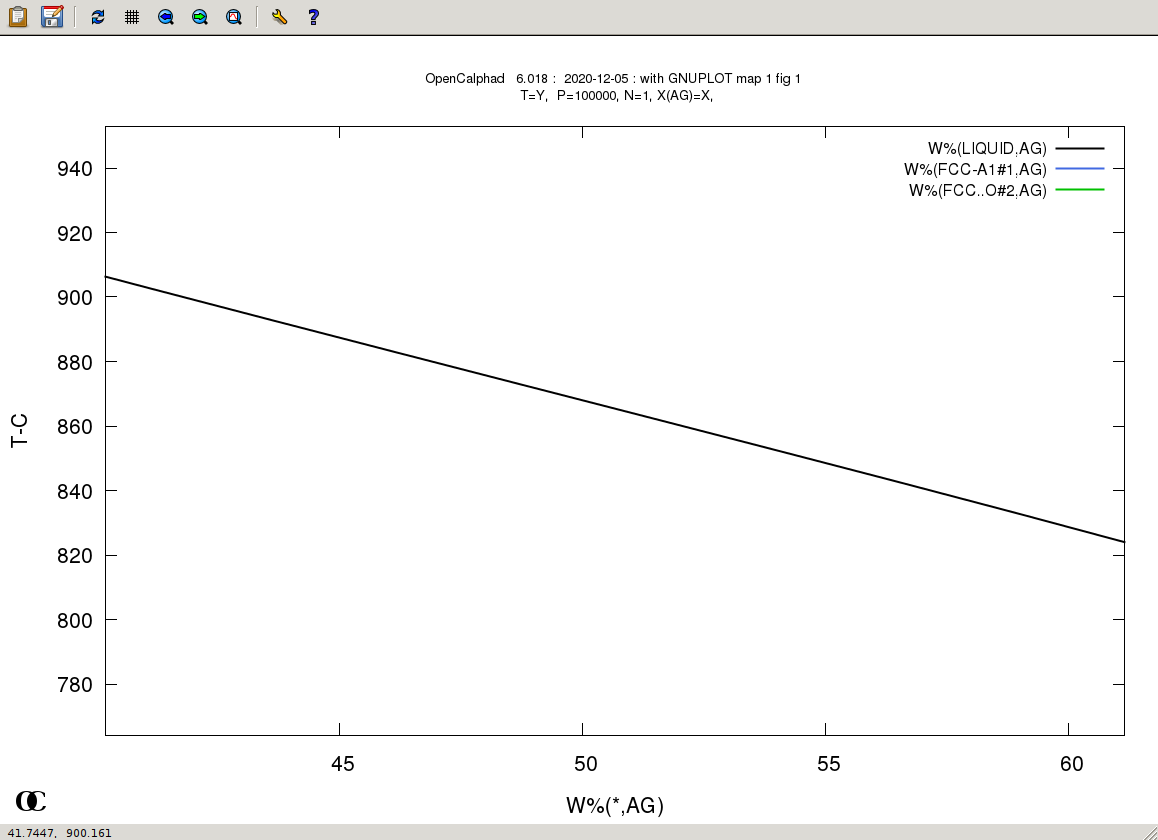
The plot can be edited and modified. Scaling x and/or y is possible. Editing the GNUPLOT plot is as well possible.

The diagram shows the use of advanced plot for specific examples.
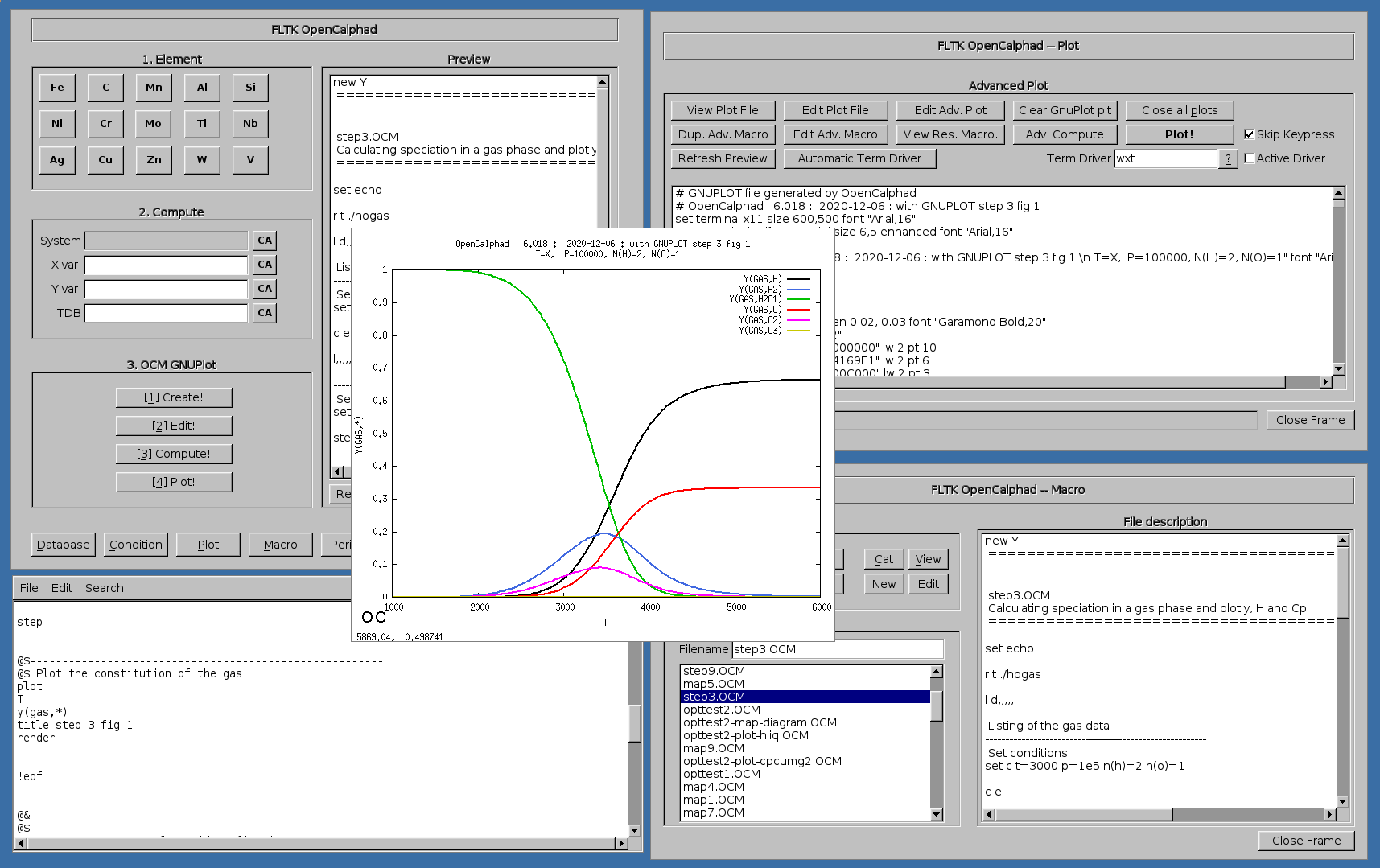
Example of calculating speciation in a gas and plot y, H and Cp:

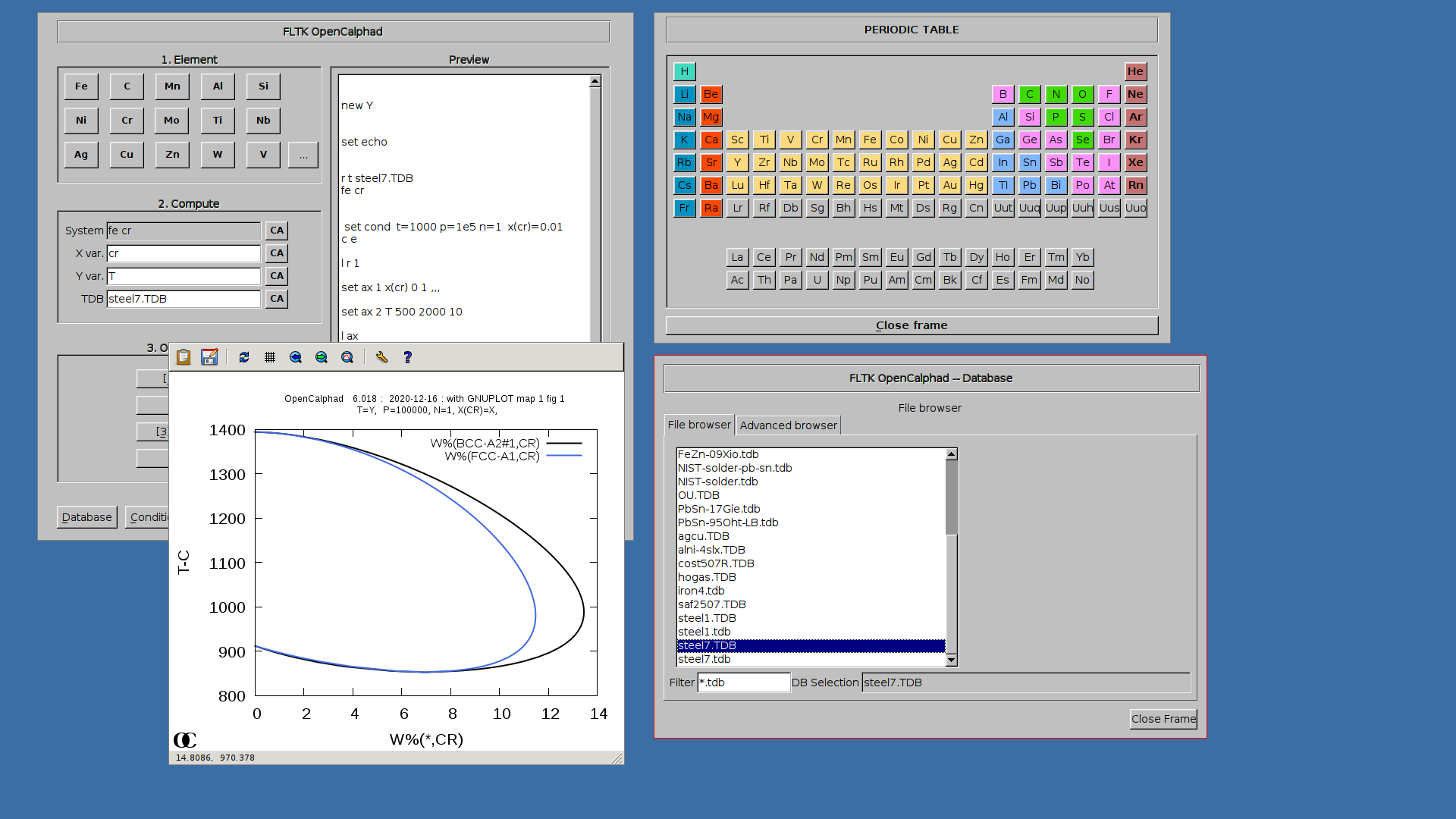
Example of OC calculation for the Cr-Fe system:
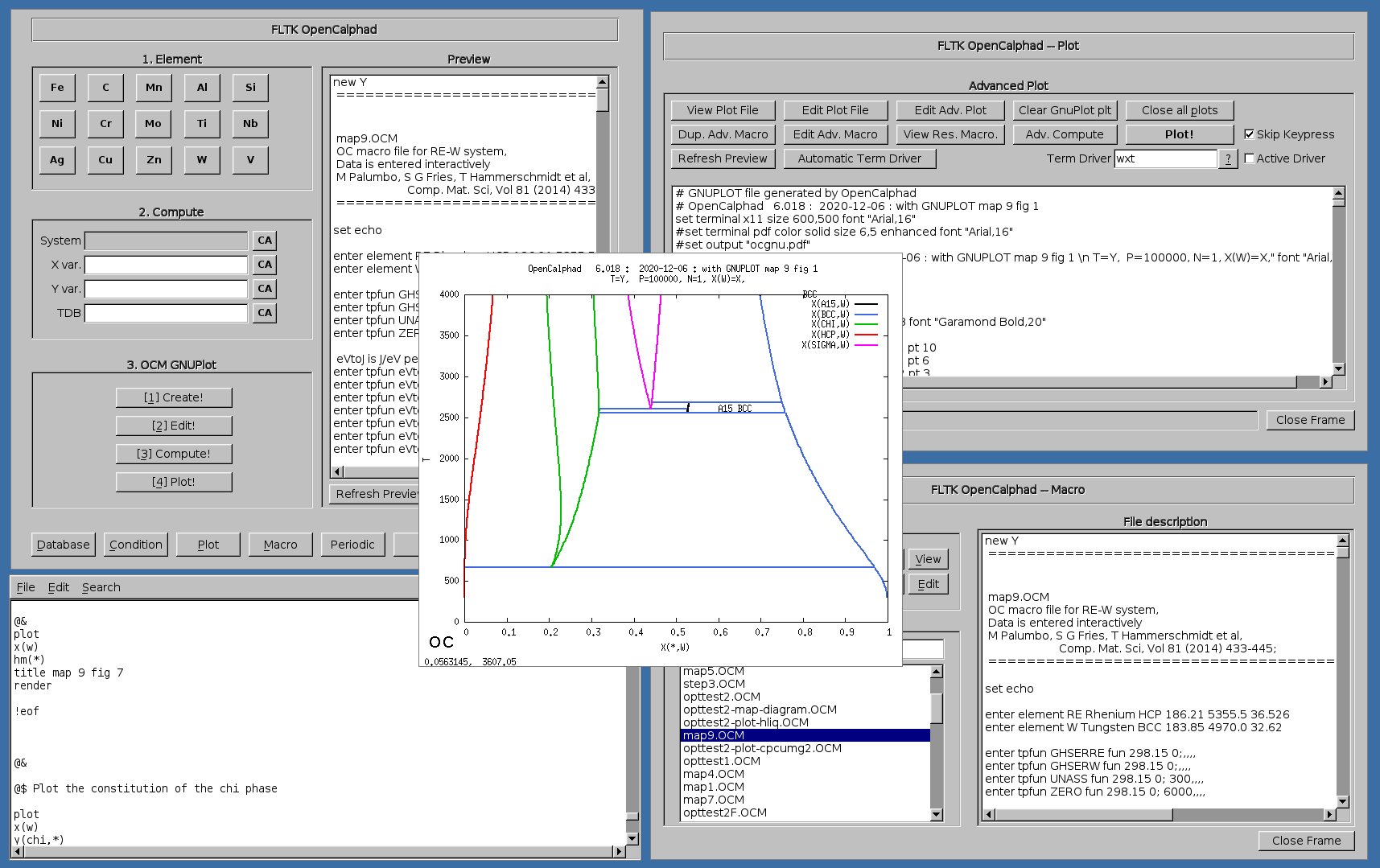
Example of OC macro file for the RE-W system (M. Palumbo, S.G. Fried, T. Hammerschmidt et al.):
Example of plot showing the property diagram for a High Speed Steel (HSS):
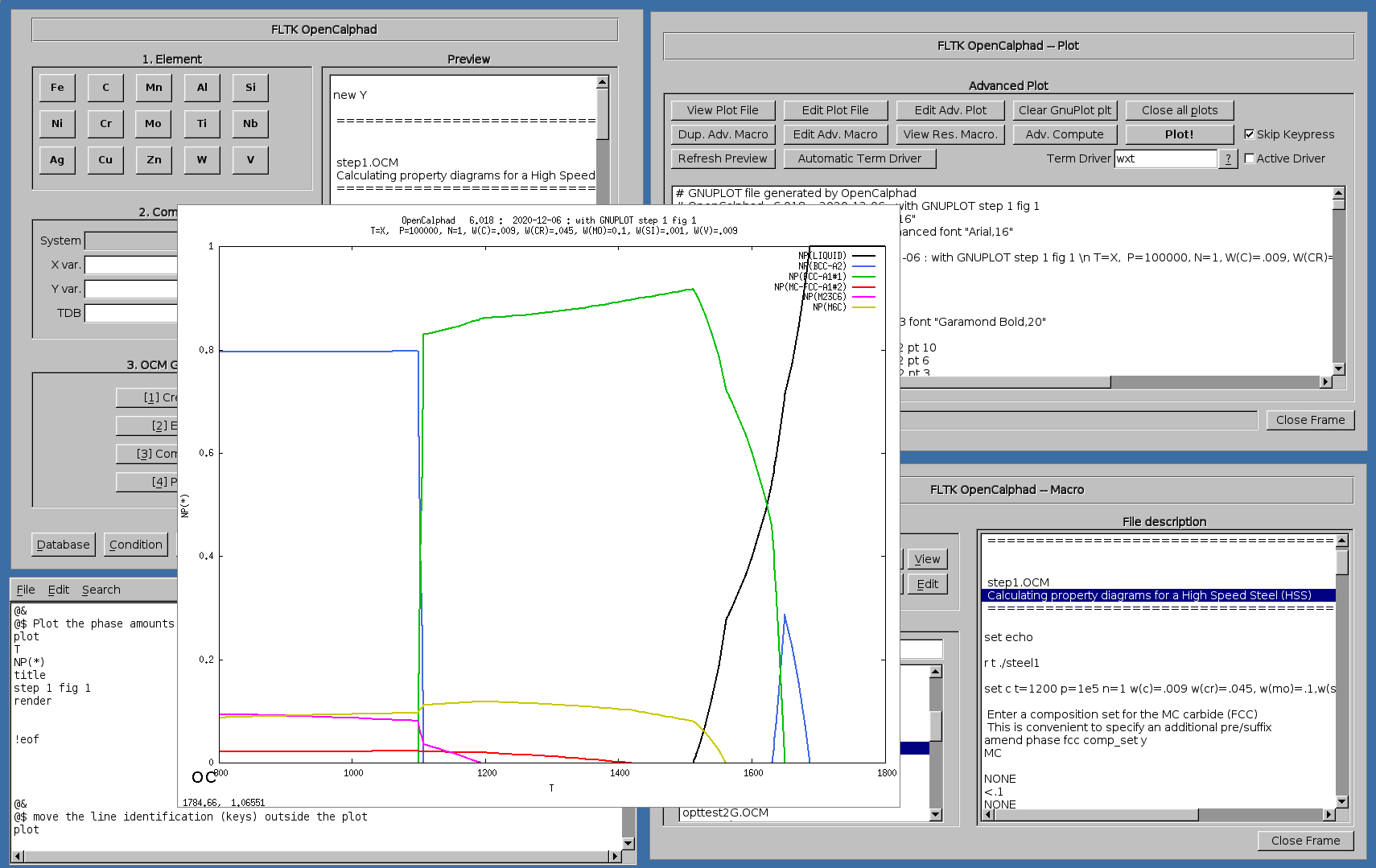
Example of plot showing the calculation of phase fractions and property diagram for SAF2507:
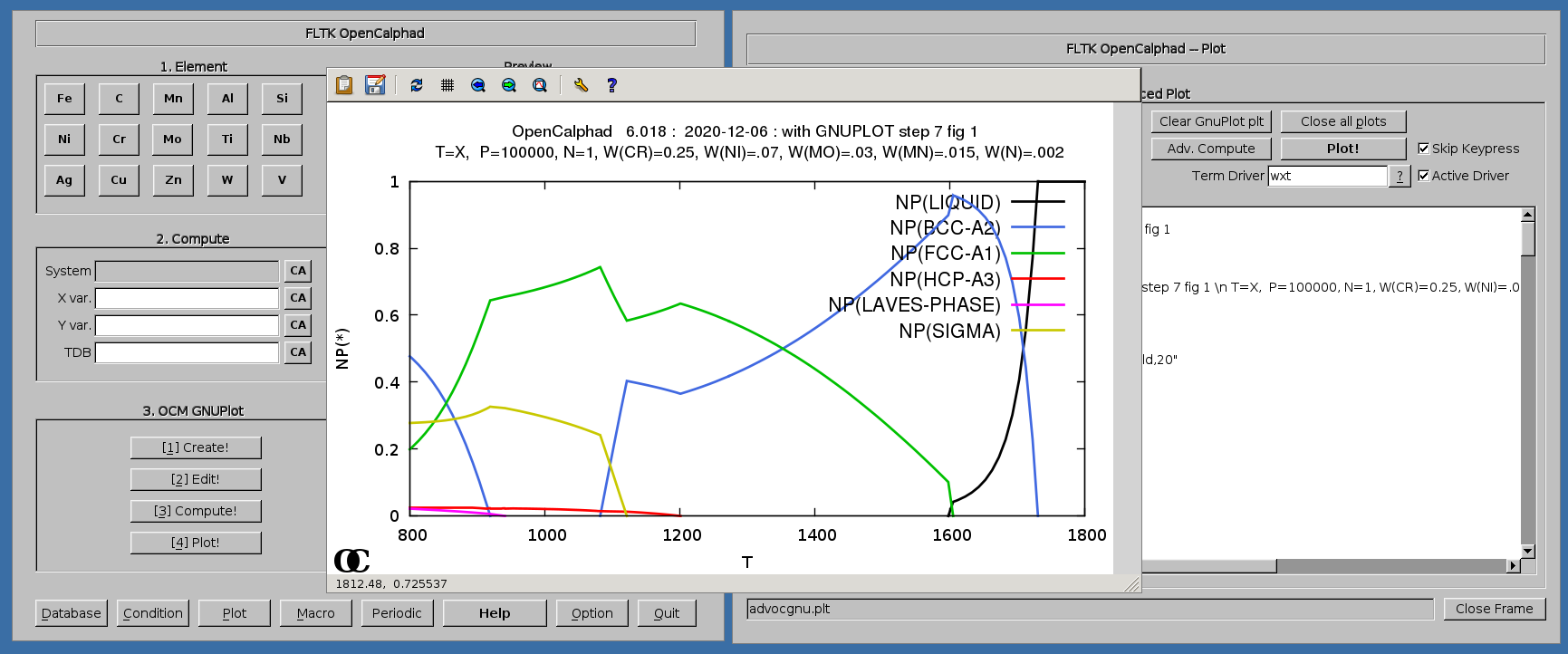
Example of ternary, calculating the isothermal section at 1200K for the Cr-Fe-Ni system:

Example of calculating G curves for the Ag-Cu system at 1000K:

Example of calculating G curves for the Fe-Mo system at 1400K:

Cu-Ni System:

Cu-Ni phase diagram calculated with OC:

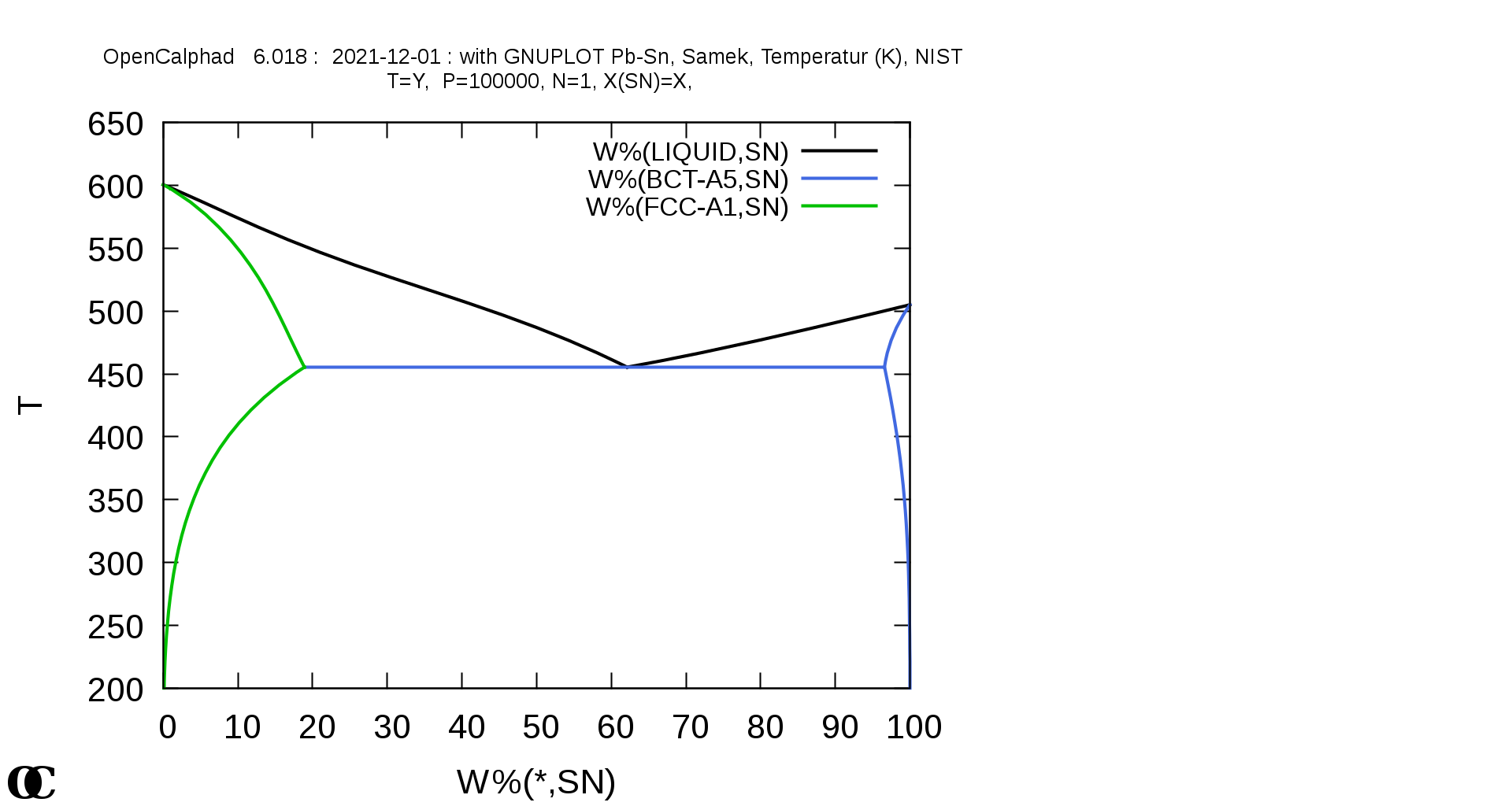
Pb-Sn phase diagram calculated with OC:










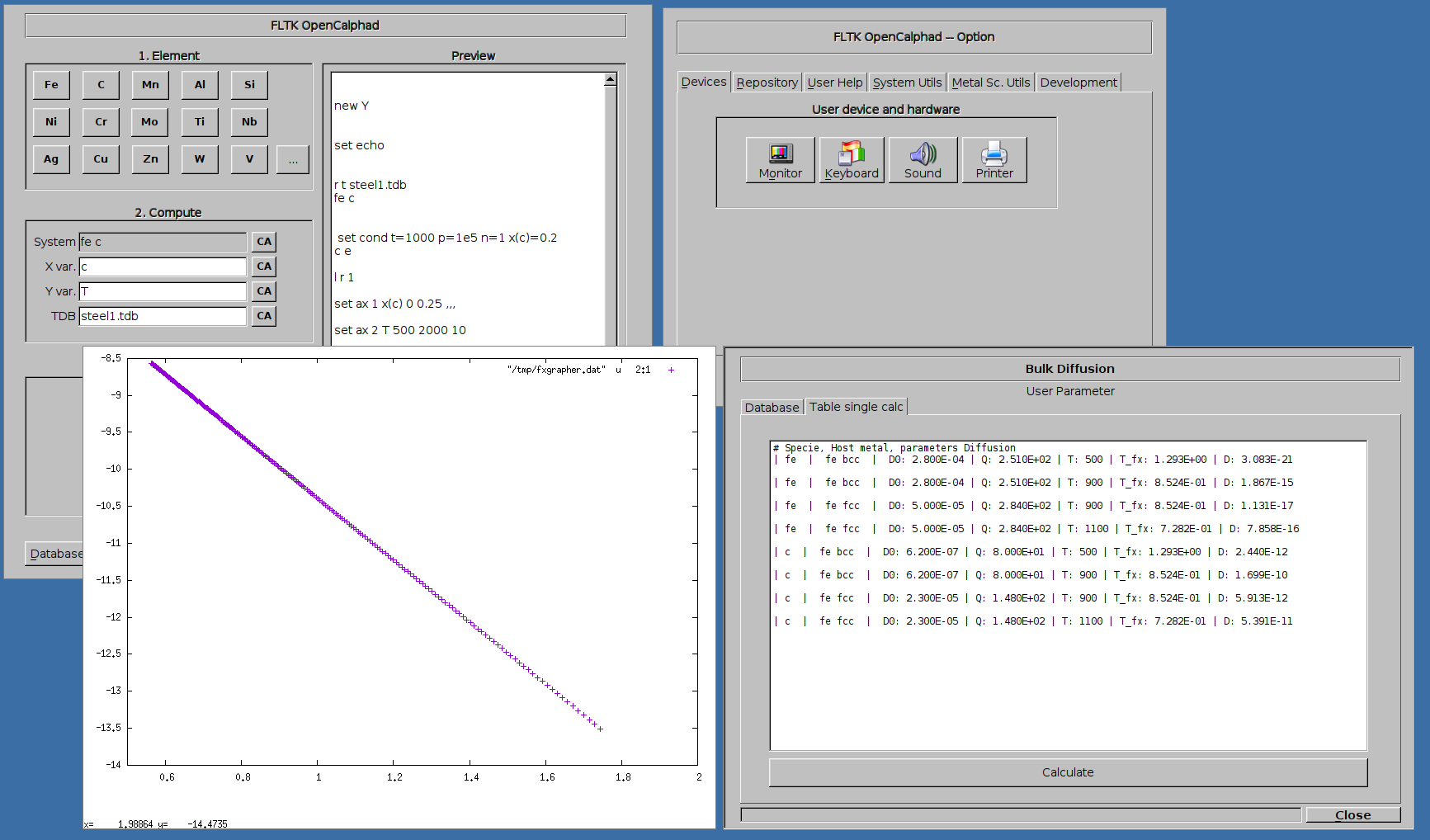
Diffusion
Several utils for materials science are included for calculation of diffusion,...
Example of plot showing the diffusion of elements (C and Fe) in host metal Fe:
The diagram for the Fe-C system can be achieved as follows.
1. Select in "Database" panel the steel1.TDB database
2. click on the button "CA" to delete the list of elements in the panel for the "System" field.
3. Click on "Fe" and "C" buttons. The set-condition field in the "Condition" panel will be proposed by the GUI as follows: " t=1000 p=1e5 n=1 x(c)=0.2 ".
4. Enter "C" for X for the simulations.
5. Click on 1.Create, 2.Edit, 3.Compute, and 4. Plot.
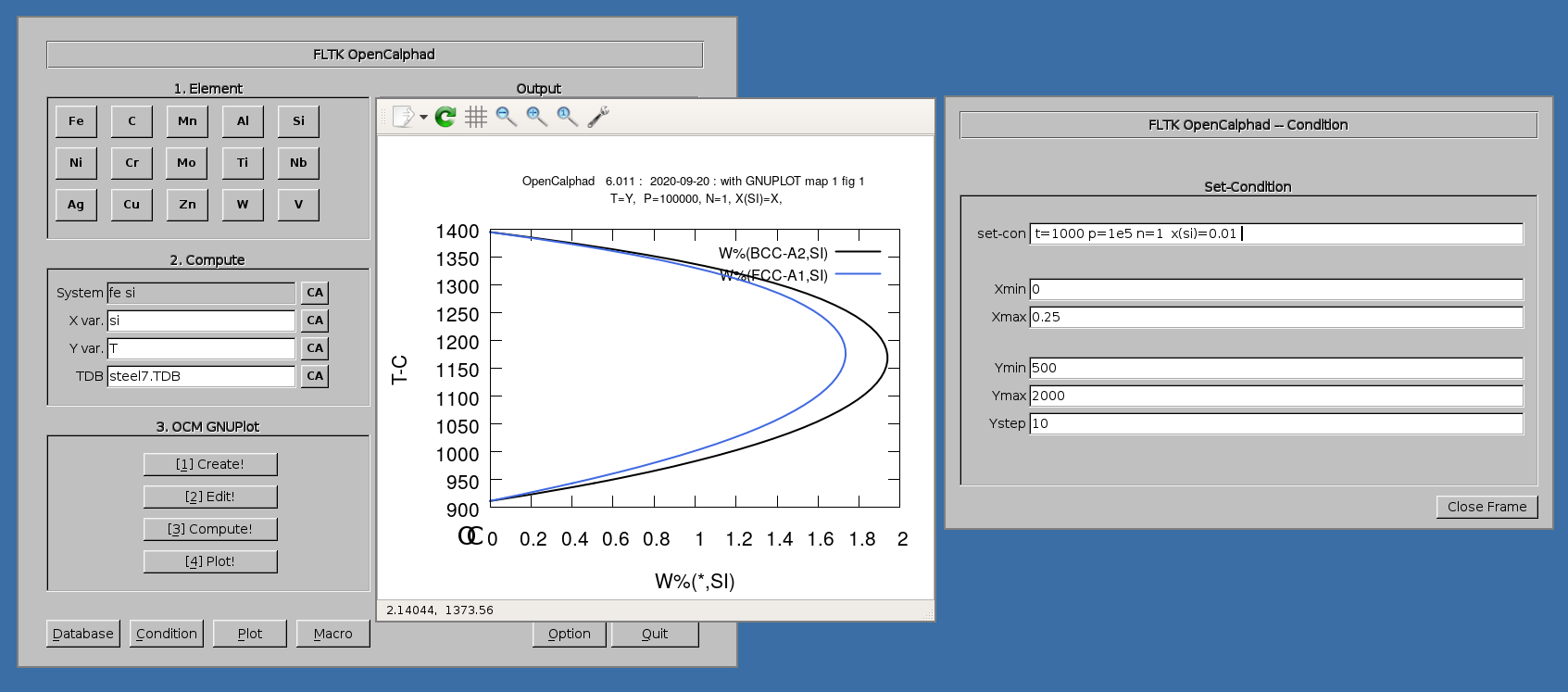
The diagram for the Fe-Si system can be achieved as follows.
1. Select in "Database" panel the steel7.TDB database
2. click on the button "CA" to delete the list of elements in the panel for the "System" field.
3. Click on "Fe" and "Si" buttons. The set-condition field in the "Condition" panel will be proposed by the GUI as follows: " t=1000 p=1e5 n=1 x(si)=0.01 ".
4. Enter "Si" for X for the simulations.
5. Click on 1.Create, 2.Edit, 3.Compute, and 4. Plot.
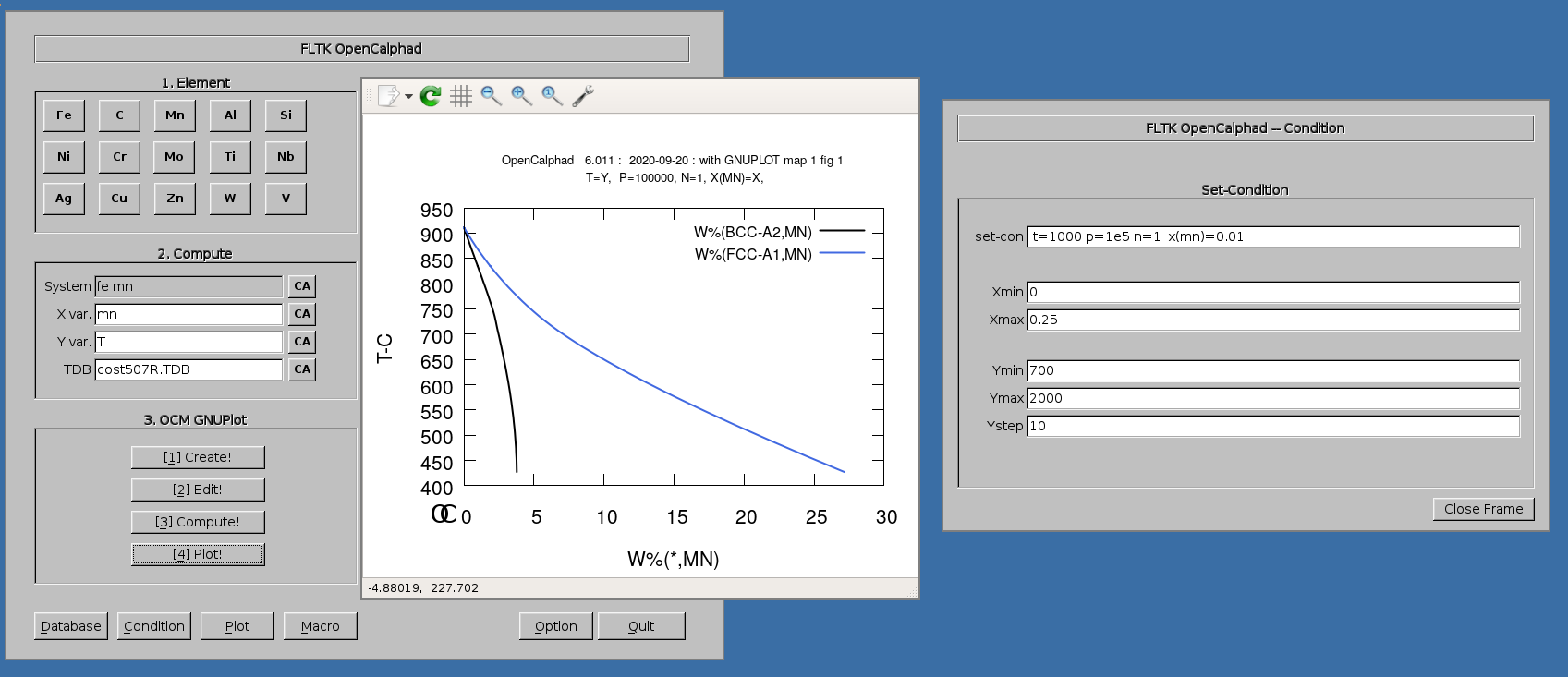
The diagram for the Fe-Mn system can be achieved as follows.
1. Select in "Database" panel the cost507R.TDB database
2. click on the button "CA" to delete the list of elements in the panel for the "System" field.
3. Click on "Fe" and "Mn" buttons. The set-condition field in the "Condition" panel will be proposed by the GUI as follows: " t=1000 p=1e5 n=1 x(mn)=0.01 ".
4. Enter "Mn" for X for the simulations.
5. Click on 1.Create, 2.Edit, 3.Compute, and 4. Plot.
The opensource graphical user interface using Linux/BSD libraries is under development. The database of OC are available on this webpage under the directory "macros" (https://github.com/lusamek/OpenCalphad/tree/master/macros). The GUI allows users to login to a platform on IRC for scientific exchanges and sharing experiences. This is avaiable in the window "Option" using the button "Development and Support".
--
January 24th, 2023:
Presentations/1674728199-1-Presentation-OpenCalphad-Mr.-Axel-Menager.pptx
IRC Channel for user exchange:
https://kiwiirc.com/nextclient/irc.libera.chat/?channels=#opencalphad
Bo Sundman, S. Fries, Chunhui Luo, B. Hallstedt, B. Grzegorczyk, A. Grajcar, A. Menager,... L.Samek.
[1] OpenCalphad Website: https://www.opencalphad.org
[2] Source Code: https://github.com/sundmanbo/opencalphad
[3] Virtualbox: https://www.virtualbox.org/
[4] https://gitlab.com/lusamek/opencalphad/ (Example, Fe-C, Fe-C-Mn,...)