GediNET- Discovering Multi-Disease Gene Associations using Established Biological Information and Knowledge-based Machine Learning
Emma Qumsiyeh1*, Louise Showe2, and Malik Yousef3,4*
1Information Technology Engineering, Al-Quds University, Palestine
2The Wistar Institute, Philadelphia, PA,19104, USA
3Department of Information Systems, Zefat Academic College, Zefat, 13206, Israel
4Galilee Digital Health Research Center (GDH), Zefat Academic College, Israel
*Corresponding authors:
Malik Yousef: malik.yousef@gmail.com
Emma Qumsiyeh: emma.qumsiyeh@hotmail.com
- Correspondence: malik.yousef@gmail.com;
To read the paper click
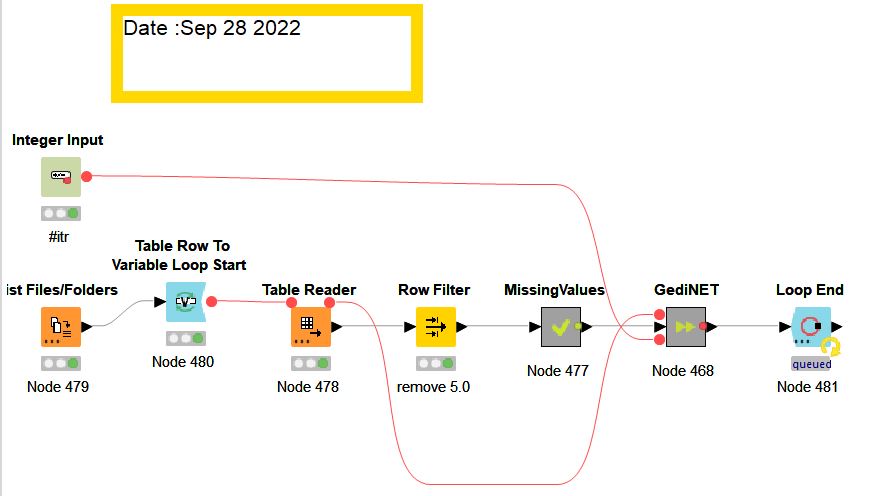
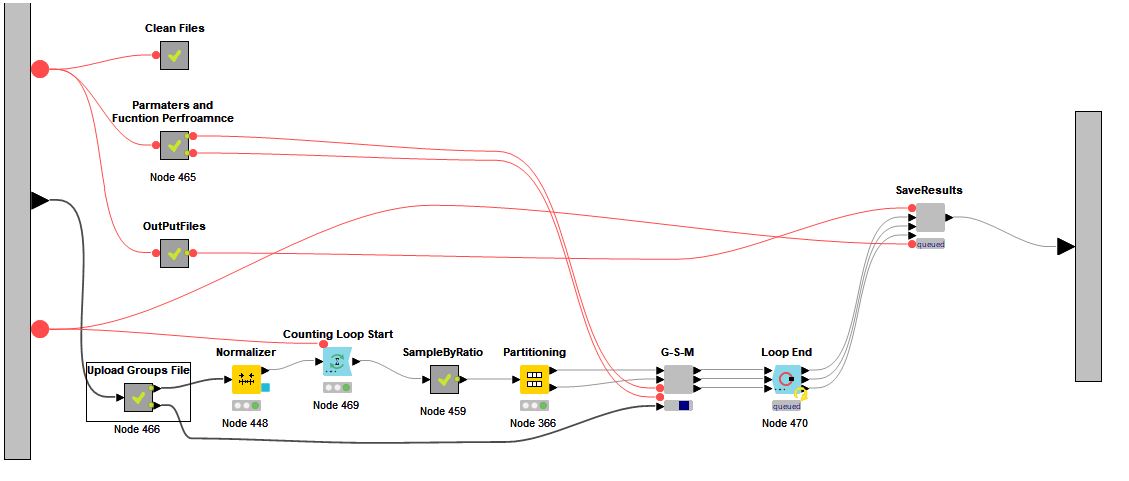
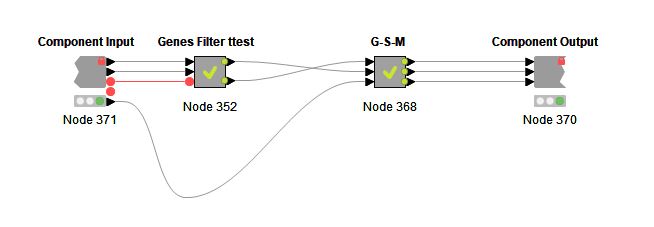
GediNET tool is a Knime workflow. In order to run the workflow, you need to download Knime and install it in your local machine.
This is the link for downloading Knime: https://www.knime.com/downloads
For more information about the Knime platform you might visit https://www.knime.com/software-overview
See this page for information about setting Knime.
Visit this page for instruction in how to prepare the dataset into Knime table format (*.table) using a Knime workflow
Visit this page for instruction in how to upload the Groups file.
Visit this page for the outputs of GediNET.
The Knime workflow name is "GediNET_v1.knwf" that you might download and run throug the Knimeplatform
Running the workflow:
- You need to use the node “MCCV Iterations” in order to specify the number of Monte Carlo Cross Validation (MCCV) iterations, for example 10 or 100.
- You need to configure the node “List Files/Folders” to point it to the folder that has the gene expression dataset in a table format (as described above)
- You might download an example of such data named DSD84.table
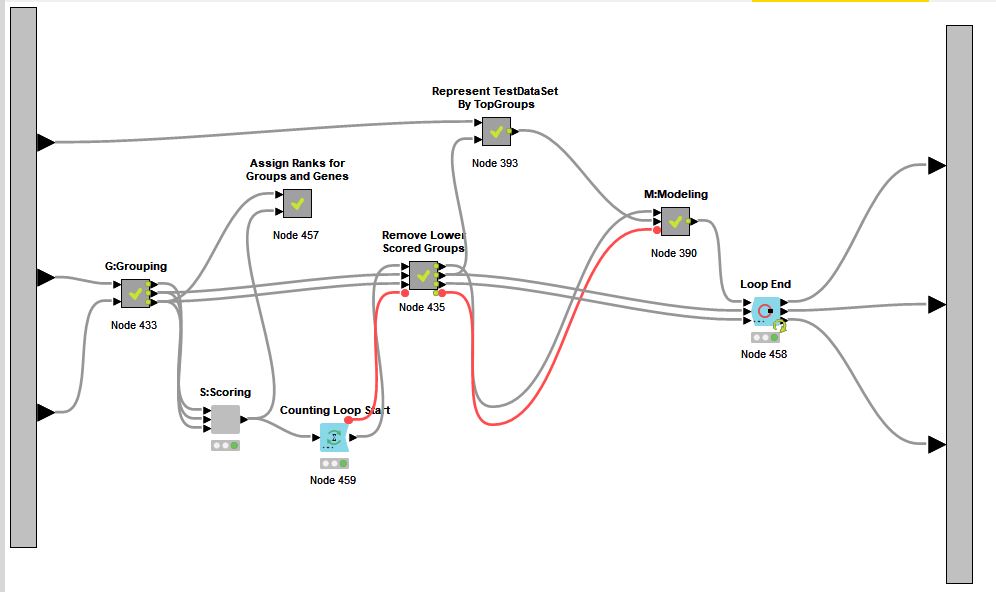
The Pseudocode of GediNET can seen over this file