
This repository provides a hands-on tensorflow implementation of Normalizing Flows as presented in the paper
introducing the concept (D. Rezende & S. Mohamed). This code was developed as part of a Special Course at DTU (Denmarks Tekniske Universitet), supervised
by Michael Riis Andersen. The final report of the course, that details all experiments run with this repository can directly be accessed at https://pierresegonne.github.io/VINF/
This repository provides an implementation of
- ADVI (Automatic Differential Variational Inference, with Diagonal Gaussian, baseline)
- Planar Flow
- Radial Flow
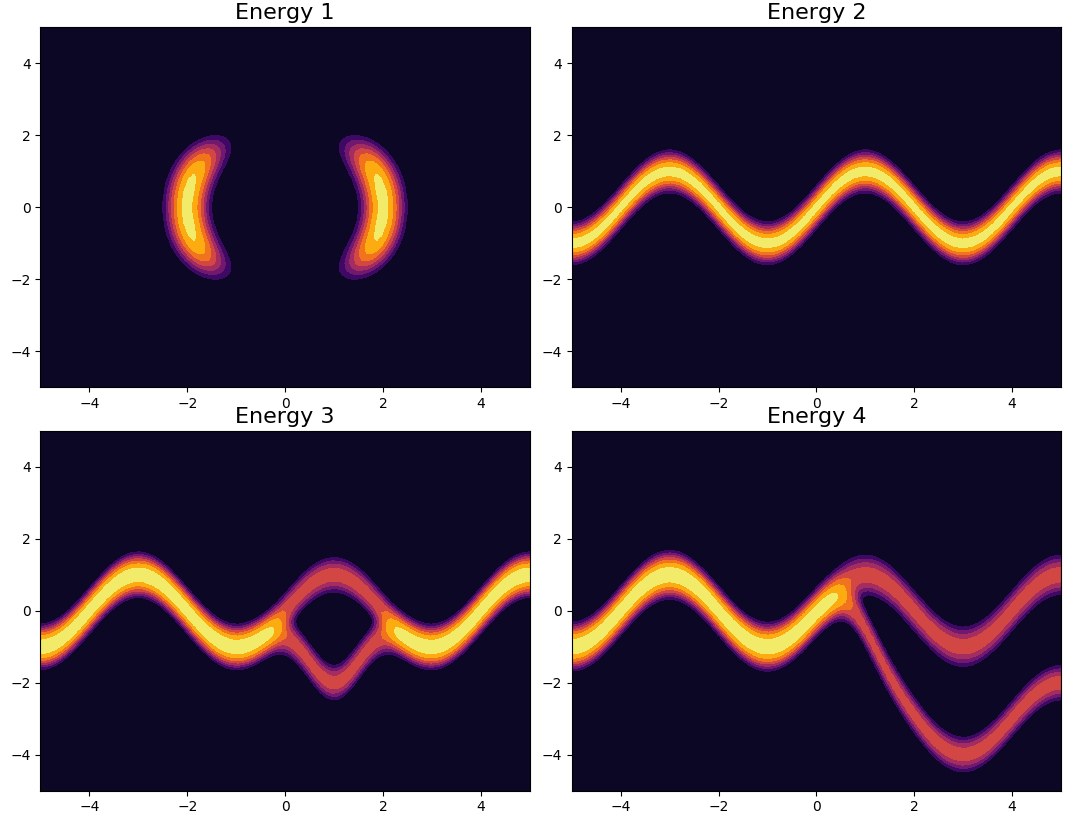
True posterior
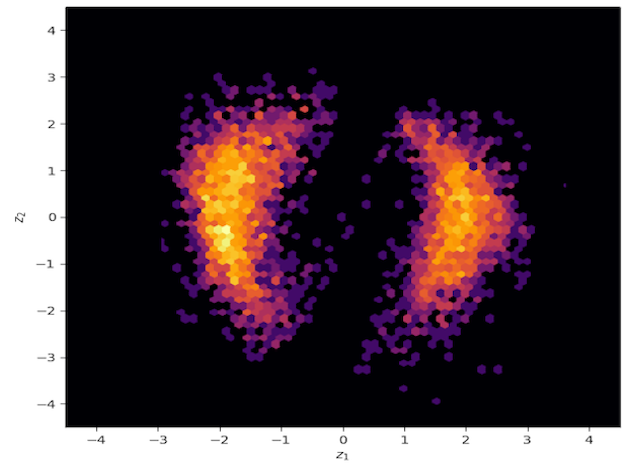
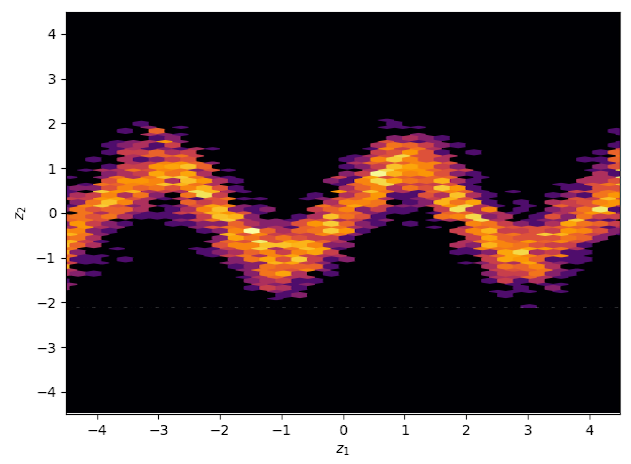
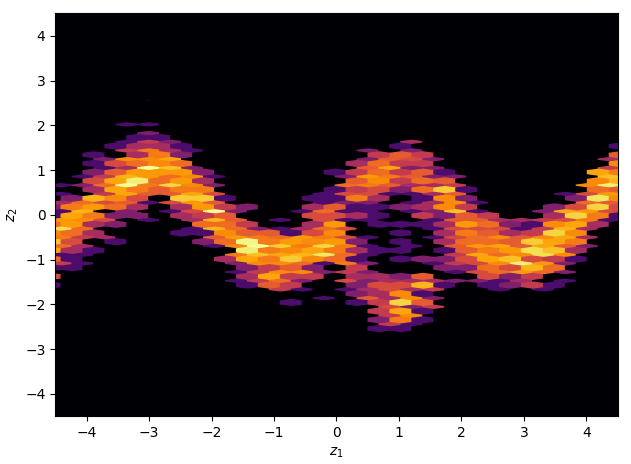
Samples generated from the trained variational approximation
- Run additional experiments on radial flows
- Add requirements.txt
- Improve models with the use of bijectors. See this thread for a starting point
- Include new flow models.