ConsensusCruncher is a tool that suppresses errors in next-generation sequencing data by using unique molecular identifers (UMIs) to amalgamate reads derived from the same DNA template into a consensus sequence.
To learn more about ConsensusCruncher and its applications: https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkz474/5498633
For a full documentation of ConsensusCruncher, please see our Read the Docs
This pipeline requires the following dependencies:
| Program | Version | Purpose |
|---|---|---|
| picrd | picard/1.9.1 or higher | Adding read group @RG in the alignment files |
| Python | 3.5.1 | Run ConsensusCruncher |
| BWA | 0.7.15 | Align reads |
| Samtools | 1.3.1 | Sorting and indexing bamfiles |
All required python libraries can be installed by running pip install -r requirements.txt
Set up config.ini with the appropriate configurations for [fastq2bam] and [consensus] modes. Alternatively, you can provide command-line arguments.
ConsensusCruncher.py processes one sample (2 paired-end FASTQ files or 1 BAM file) at a time. A sample script to generate shell scripts for multiple samples is available here.
- Run ConsensusCruncher.py [-c CONFIG] fastq2bam with required input parameters:
--fastq1 FASTQ1 FASTQ containing Read 1 of paired-end reads. [MANDATORY]
--fastq2 FASTQ2 FASTQ containing Read 2 of paired-end reads. [MANDATORY]
-o OUTPUT, --output OUTPUT
Output directory, where barcode extracted FASTQ and
BAM files will be placed in subdirectories 'fastq_tag'
and 'bamfiles' respectively (dir will be created if
they do not exist). [MANDATORY]
-n FILENAME, --name FILENAME
Output filename. If none provided, default will
extract output name by taking everything left of '_R'.
-b BWA, --bwa BWA Path to executable bwa. [MANDATORY]
-r REF, --ref REF Reference (BWA index). [MANDATORY]
-s SAMTOOLS, --samtools SAMTOOLS
Path to executable samtools [MANDATORY]
-p PATTERN, --bpattern PATTERN
Barcode pattern (N = random barcode bases, A|C|G|T =
fixed spacer bases). [Pattern or list must be provided]
-l LIST, --blist LIST
List of barcodes (Text file with unique barcodes on
each line). [Pattern or list must be provided]
BARCODE DESIGN: You can input either a barcode list or barcode pattern or both. If both are provided, barcodes will first be matched with the list and then the constant spacer bases will be removed before the barcode is added to the header. N = random / barcode bases A | C | G | T = constant spacer bases e.g. ATNNGT means barcode is flanked by two spacers matching 'AT' in front and 'GT' behind.
DESCRIPTION: This script extracts molecular barcode tags and removes spacers from unzipped FASTQ files found in the input directory (file names must contain "R1" or "R2"). Barcode extracted FASTQ files are written to the 'fastq_tag' directory and are subsequently aligned with BWA mem. Bamfiles are written to the 'bamfile" directory under the project folder.
- Run ConsensusCruncher.py [-c CONFIG] consensus with the required input parameters:
-h, --help show this help message and exit
-i BAM, --input BAM Input BAM file with barcodes extracted into header. [mandatory]
-o OUTPUT, --output OUTPUT
Output directory, where a folder will be created for
the BAM file and consensus sequences. [mandatory]
-s SAMTOOLS, --samtools SAMTOOLS
Path to executable samtools. [mandatory]
--scorrect {True,False}
Singleton correction, default: True.
-b BEDFILE, --bedfile Separator file to split bamfile into chunks for processing.
Default: hg19 cytoband (You can find other cytobands for your
genome of interest on UCSC
http://hgdownload.cse.ucsc.edu/downloads.html).
For small BAM files, you may choose to turn off data splitting
with '-b False' and process everything all at once (Division of
data is only required for large data sets to offload the
memory burden).
--cutoff CUTOFF Consensus cut-off, default: 0.7 (70% of reads must
have the same base to form a consensus).
--cleanup {True,False}
Remove intermediate files.
This script amalgamates duplicate reads in bamfiles into single-strand consensus sequences (SSCS), which are subsequently combined into duplex consensus sequences (DCS). Singletons (reads lacking duplicate sequences) are corrected, combined with SSCS to form SSCS + SC, and further collapsed to form DCS + SC. Finally, files containing all unique molecules (a.k.a. no duplicates) are created for SSCS and DCS.
script generator will create sh scripts for each file in a fastq directory.
- The following parameters need to be changed in the config file: name, bwa, ref, samtools, bpattern (alternatively if a barcode list is used instead, remove bpattern and add blist as parameter). Please note: fastq1, fastq2, output, bam, and c_output can be ignored as those will be updated using the generate_scripts.sh file.
- Update generate_scripts.sh with input, output, and code_dir.
- Run generate_scripts.sh to create sh files and then run those scripts.
In order to create consensus sequences, we first need to process fastq files into bam files. Sample fastq files can be found under the test folder. Please note these fastqs are only for testing purposes. For the full fastqs used in our paper, please download the data from the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra/) under access numbers SRP140497 and SRP141184.
Given fastq as input files, fastq2bam mode removes the spacer region and extracts the barcode tag from each sequencing read into the header with extract_barcode.py.
REPO="[insert path to ConsensusCruncher repo]"
BWAPATH="[insert path to BWA]"
BWAINDEX="[insert path to BWA INDEX]"
BWAPATH="[insert path to SAMTOOLS]"
python ConsensusCruncher.py fastq2bam --fastq1 $REPO/test/fastq/LargeMid_56_L005_R1.fastq --FASTQ2 $REPO/test/fastq/LargeMid_56_L005_R2.fastq -o $REPO/test -b $BWAPATH -r $BWAIndex -s $SAMTOOLS -bpattern NNT
In the sample dataset, we utilized 2-bp (NN) barcodes and 1-bp (T) spacers. While the barcodes for each read can be one of 16 possible combinations (4^2), the spacer is an invariant "T" base used to ligate barcodes onto each end of a DNA fragment. Thus, a spacer filter is imposed to remove faulty reads. Barcodes from read 1 and read 2 are extracted and combined together before being added to the header.
READ FROM SEQUENCER
Read1:
@HWI-D00331:196:C900FANXX:5:1101:1332:2193 1:N:0:ACGTCACA [<-- HEADER]
ATTAAGCCCCAGGCAGTTGCTAATGATGGGAGCTTAGTGCACAAGGGCTGGGCCTCCCTCTTGGAGCTGAACATTGTTTCTTGGGGACGGCTGTGCCCACCTCAGCGGGGAGGCAAGGATTAAATC [<-- SEQUENCE]
+
BCCCCGGGGGGGGGGGGGGGGGGGGGGGGGFGGGGGGGGEGGGGGBGGGGGGGGGGGGGGGGGGGGGGGEGG1:FGFGGGGGGGGG/CB>DG@GGGGGGG<DGGGGAAGGEGGB>DGGGEGGG/@G [<-- QUALITY SCORE]
Read2:
@HWI-D00331:196:C900FANXX:5:1101:1332:2193 2:N:0:ACGTCACA
GGTGGGCTCCAGCCCTGATTTCCTCCCCCAGCCCTGCAGGGCTCAGGTCCAGAGGACACAAGTTTAACTTGCGGGTGGTCACTTGCCTCGTGCGGTGACGCCATGGTGCCCTCTCTGTGCAGCGCA
+
BBBBCGGGGEGGGGFGGGGGGGGGGGGGGGGGGGGGGB:FCGGGGGGGGGGEGGGGGGGG=FCGG:@GGGEGBGGGAGFGDE@FGGGGGFGFGEGDGGGFCGGDEBGGGGGGGEG=EGGGEEGGG#
------
AFTER BARCODE EXTRACTION AND SPACER ("T") REMOVAL
Read1:
@HWI-D00331:196:C900FANXX:5:1101:1332:2193|ATGG/1
AAGCCCCAGGCAGTTGCTAATGATGGGAGCTTAGTGCACAAGGGCTGGGCCTCCCTCTTGGAGCTGAACATTGTTTCTTGGGGACGGCTGTGCCCACCTCAGCGGGGAGGCAAGGATTAAATC
+
CCGGGGGGGGGGGGGGGGGGGGGGGGGFGGGGGGGGEGGGGGBGGGGGGGGGGGGGGGGGGGGGGGEGG1:FGFGGGGGGGGG/CB>DG@GGGGGGG<DGGGGAAGGEGGB>DGGGEGGG/@G
Read2:
@HWI-D00331:196:C900FANXX:5:1101:1332:2193|ATGG/2
GGGCTCCAGCCCTGATTTCCTCCCCCAGCCCTGCAGGGCTCAGGTCCAGAGGACACAAGTTTAACTTGCGGGTGGTCACTTGCCTCGTGCGGTGACGCCATGGTGCCCTCTCTGTGCAGCGCA
+
BCGGGGEGGGGFGGGGGGGGGGGGGGGGGGGGGGB:FCGGGGGGGGGGEGGGGGGGG=FCGG:@GGGEGBGGGAGFGDE@FGGGGGFGFGEGDGGGFCGGDEBGGGGGGGEG=EGGGEEGGG#
FASTQ files with extracted barcodes are placed in the fastq_tag directory and are subsequently aligned with BWA to generate BAMs in the bamfiles folder.
.
├── bamfiles
├── fastq
├── fastq_tag
└── qsub
consensus mode creates a consensus directory and folders for each bam file.
BAM files undergo consensus construction through the workflow illustrated above. Output BAMs are grouped according to type of error suppression (SSCS vs DCS) and whether Singleton Correction (SC) was implemented.
.
├── bamfiles
├── consensus
│ ├── LargeMid_56_L005
│ │ ├── dcs
│ │ ├── dcs_SC
│ │ ├── sscs
│ │ └── sscs_SC
...
│ ├── LargeMid_62_L006
│ │ ├── dcs
│ │ ├── dcs_SC
│ │ ├── sscs
│ │ └── sscs_SC
│ └── qsub
├── fastq
├── fastq_tag
└── qsub
Within a sample directory (e.g. LargeMid_56_L005), you will find the following files:
Please note the example below is for illustrative purposes only, as sample names and index files were removed for simplification. Order of directories and files were also altered to improve comprehension.
. Filetype
├── sscs
│ ├── badReads.bam Reads that are unmapped or have multiple alignments
│ ├── sscs.sorted.bam Single-Strand Consensus Sequences (SSCS)
│ ├── singleton.sorted.bam Single reads (Singleton) that cannot form SSCSs
├── sscs_SC
| ├── singleton.rescue.sorted.bam Singleton correction (SC) with complementary singletons
| ├── sscs.rescue.sorted.bam SC with complementary SSCSs
| ├── sscs.sc.sorted.bam SSCS combined with corrected singletons (from both rescue strategies) [*]
| ├── rescue.remaining.sorted.bam Singletons that could not be corrected
| ├── all.unique.sscs.sorted.bam SSCS + SC + remaining (uncorrected) singletons
├── dcs
│ ├── dcs.sorted.bam Duplex Consensus Sequence (DCS)
│ ├── sscs.singleton.sorted.bam SSCSs that could not form DCSs as complementary strand was missing
├── dcs_SC
│ ├── dcs.sc.sorted.bam DCS generated from SSCS + SC [*]
│ ├── sscs.sc.singleton.sorted.bam SSCS + SC that could not form DCSs
│ ├── all.unique.dcs.sorted.bam DCS (from SSCS + SC) + SSCS_SC_Singletons + remaining singletons
├── read_families.txt Family size and frequency
├── stats.txt Consensus sequence formation metrics
├── tag_fam_size.png Distribution of reads across family size
└── time_tracker.txt Time log
Through each stage of consensus formation, duplicate reads are collapsed together and single reads are written as separate files. This allows rentention of all unique molecules, while providing users with easy data management for cross-comparisons between error suppression strategies.
To simplify analyses, it would be good to focus on SSCS+SC ("sscs.sc.sorted.bam") and DCS+SC ("dcs.sc.sorted.bam") as highlighted above with [*].
Unique molecular identifiers (UMIs) composed of molecular barcodes and sequence features are used aggregate reads derived from the same strand of a template molecule. Amalgamation of such reads into single strand consensus sequences (SSCS) removes discordant bases, which effectively eliminates polymerase and sequencer errors. Complementary SSCSs can be subsequently combined to form a duplex consensus sequence (DCS), which eliminates asymmetric strand artefacts such as those that develop from oxidative damage.
Conventional UMI-based strategies rely on redundant sequencing from both template strands to form consensus sequences and cannot error suppress single reads (singleton). We enable singleton correction using complementary duplex reads in the absence of redundant sequencing.
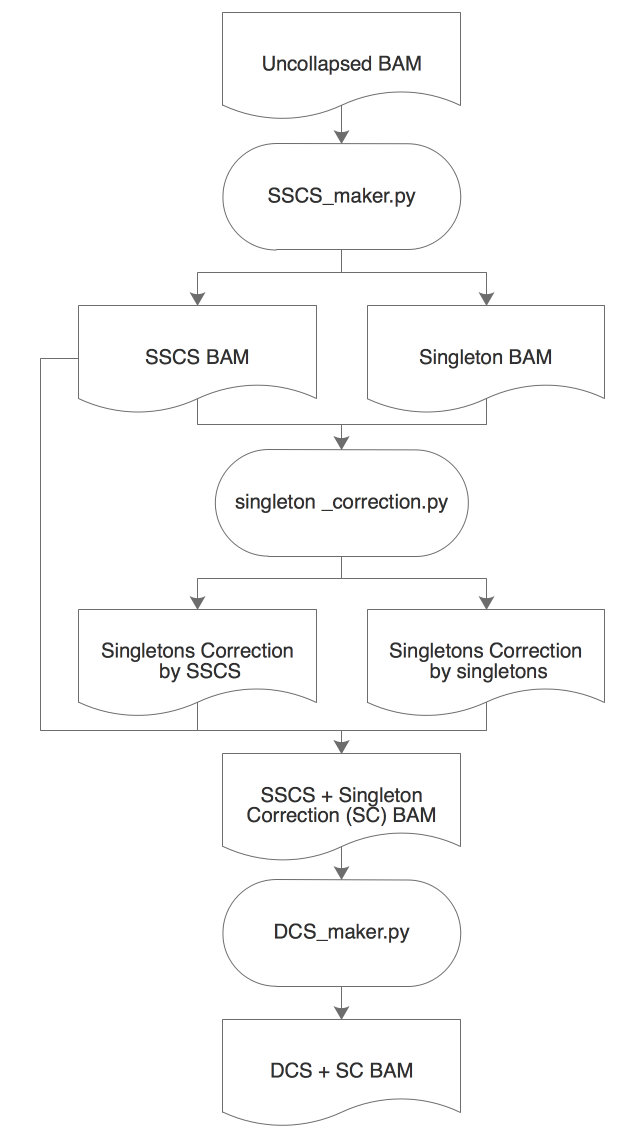
ConsensusCruncher schematic:
- An uncollapsed bamfile is first processed through SSCS_maker.py to create an error-suppressed single-strand consensus sequence (SSCS) bamfile and an uncorrected singleton bamfile.
- The singletons can be corrected through singleton_correction.py, which error suppress singletons with its complementary SSCS or singleton read.
- SSCS reads can be directly made into duplex consensus sequences (DCS) or merged with corrected singletons to create an expanded pool of DCS reads (Figure illustrates singleton correction merged work flow).
Please use this repository templates available at .github/ISSUE_TEMPLATE
- Jeff Bruce (Jeffrey.Bruce@uhnresearch.ca), Trevor Pugh (Trevor.Pugh@uhn.ca), Scott Bratman (Scott.Bratman@rmp.uhn.ca)