
Key Features • How To Use • Credits • License

-
This machine learning model takes a RNA sequence and predicts what class does it belong to. Classes are taken as taxonomies. The avaible taxonomies are the following 19:
- Orthomyxoviridae
- Rhabdoviridae
- Arteriviridae
- Coronaviridae
- Reoviridae
- Caliciviridae
- Phenuiviridae
- Hantaviridae
- Picornaviridae
- Betaflexiviridae
- Astroviridae
- Closteroviridae
- Flaviviridae
- Potyviridae
- Retroviridae
- Togaviridae
- Paramyxoviridae
- Hepeviridae
- Pneumoviridae
-
Before Prediction the model computes a markov chain whose states are the 64 writeable codons with the nucleoids A, C, G, T and then computes metrics over its adjacent associated matrix: 8 of them are matricial norms and the missing 10 parameters are the first eigenvalues complex norms ascending ordered. Namely:
Frobenius NormNuclear NormInfty NormNeg Infty NormNeg L1 NormL1 NormNeg L2 NormL2 Normeig 1eig 2eig 3eig 4eig 5eig 6eig 7eig 8eig 9eig 10
With these new metrics, we built a new dataset. and we found this scatter plot:

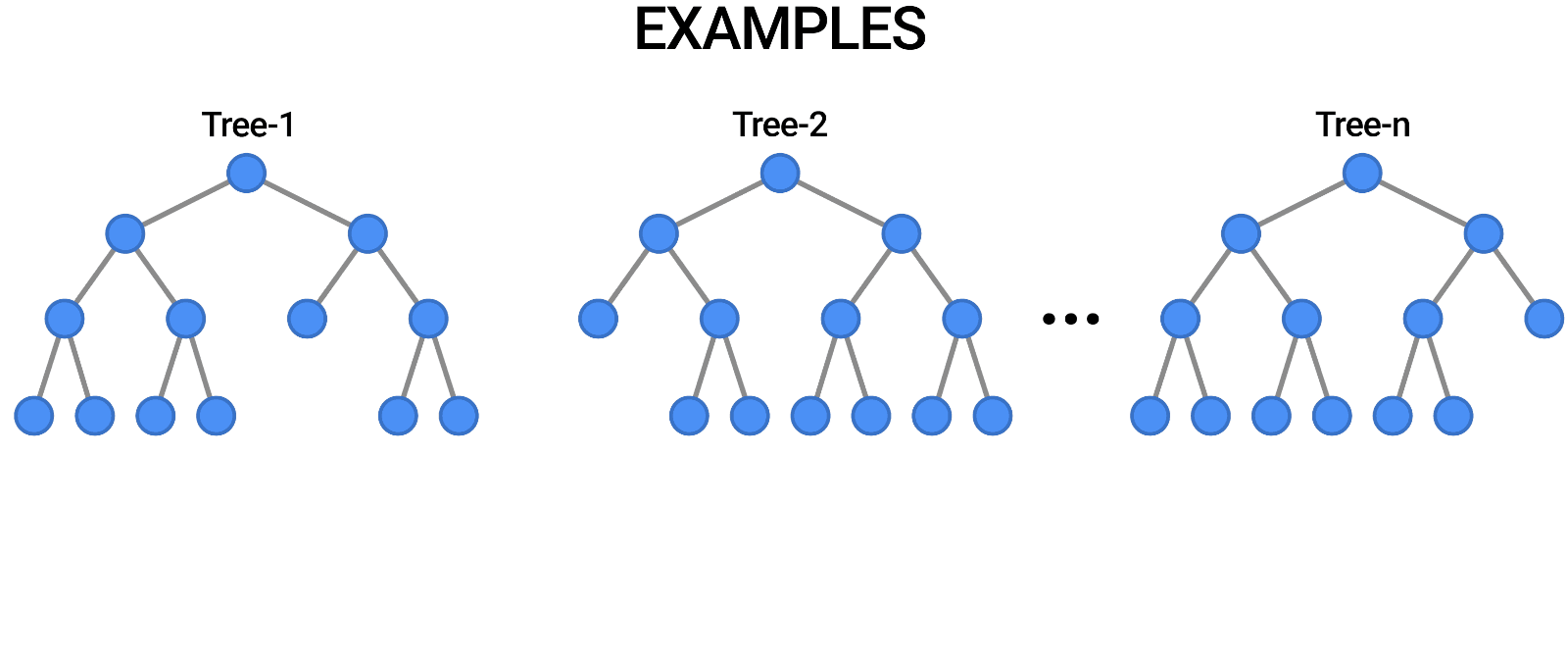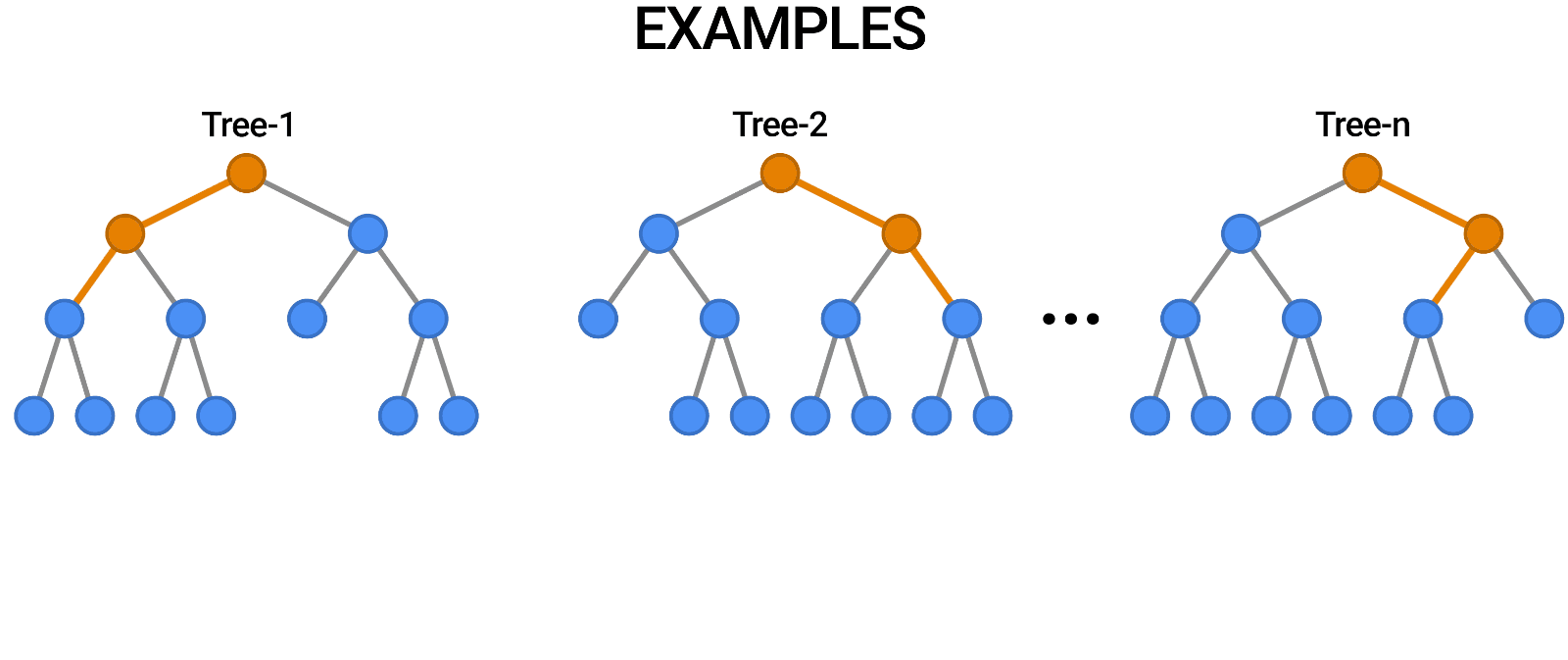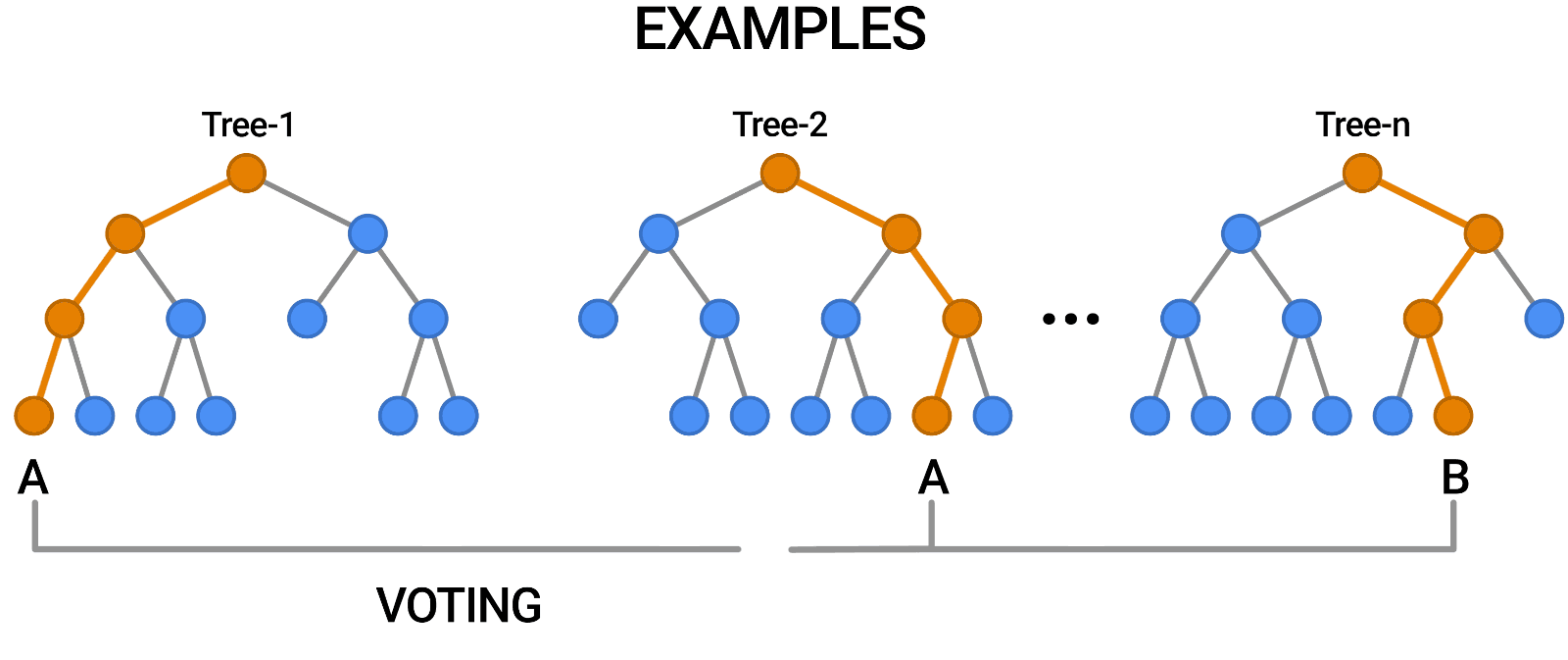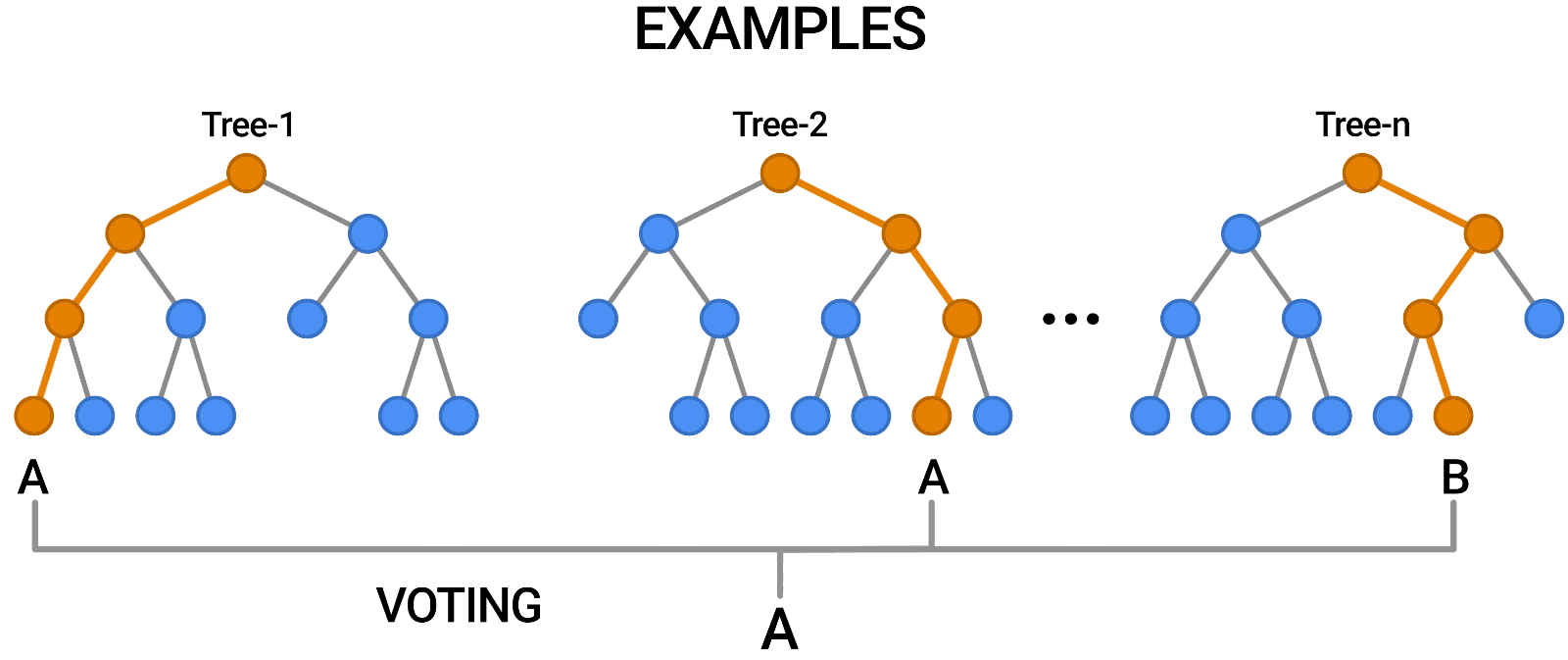
- We implemented a Random Forest model whose train data is taken from the new dataset.
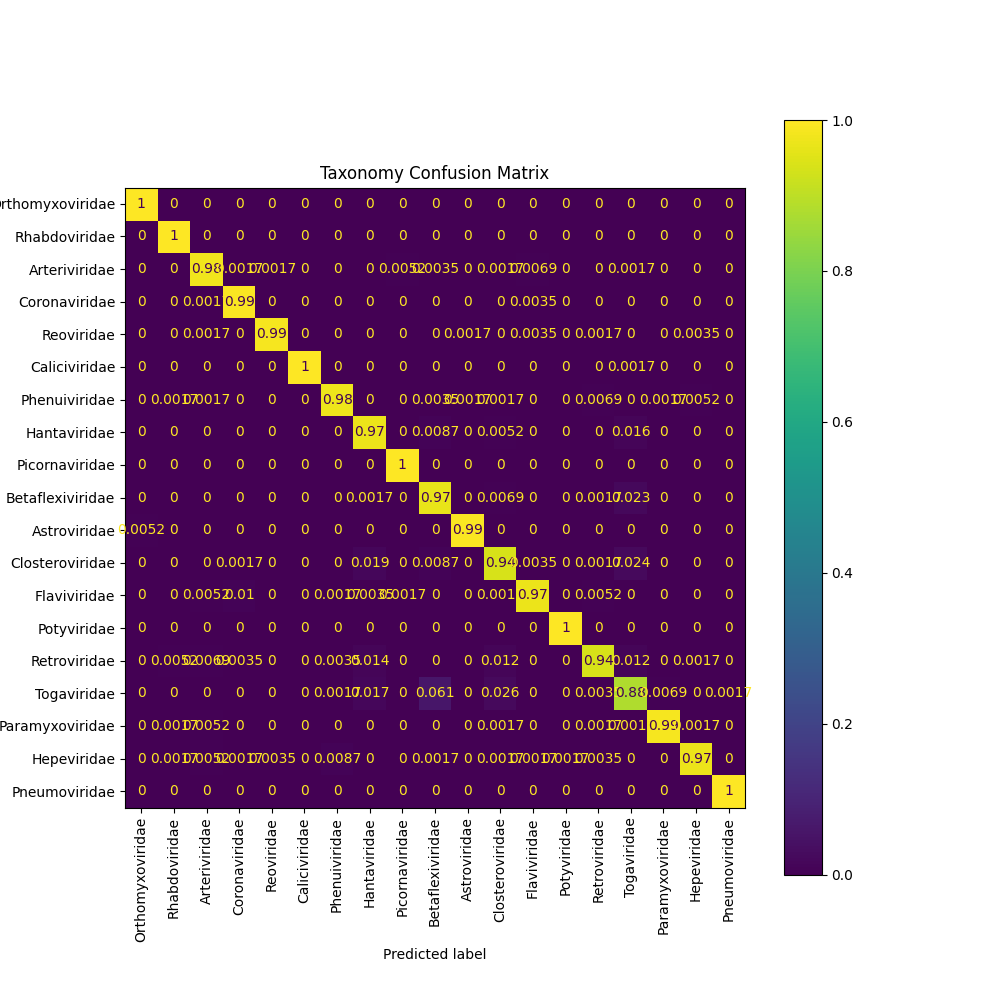
- We archieved a 96.9% of F1 score on validation set.
- The confusion matrix is the following
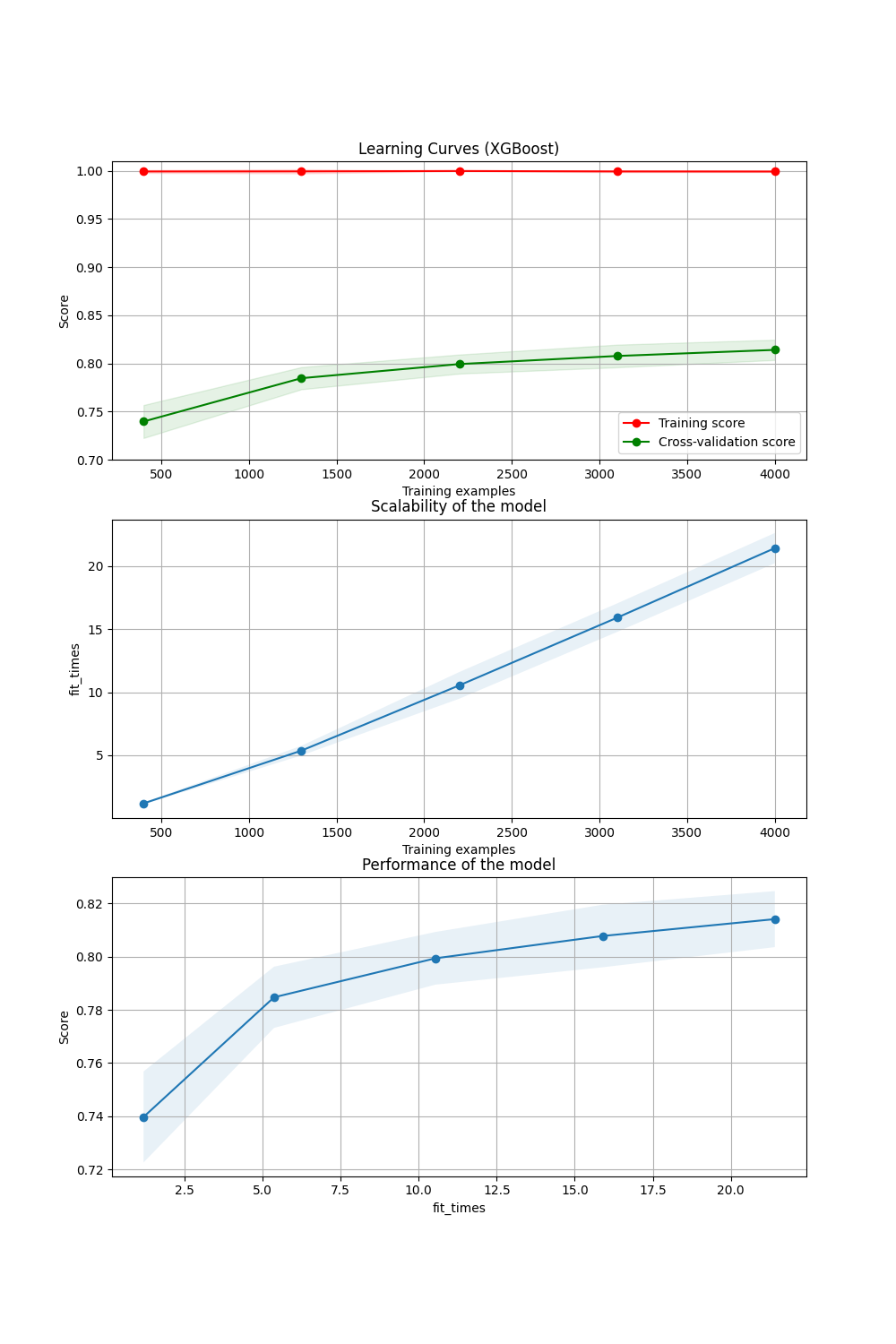
- The learning curve is the following

To clone and run this application, follow these steps
# Clone this repository
$ git clone https://github.com/santiagoahl/rna-taxonomy-prediction.git
# Go into the repository
$ cd rna-taxonomy-prediction
# Go to jupyter notebooks
$ jupyter-notebook
# Run the Libraries & Modules cell
# Run the Model Import cellThis software uses the following packages:
MIT
Web Site santiagoal.super.site · GitHub @santiagoahl · Twitter @sahumadaloz