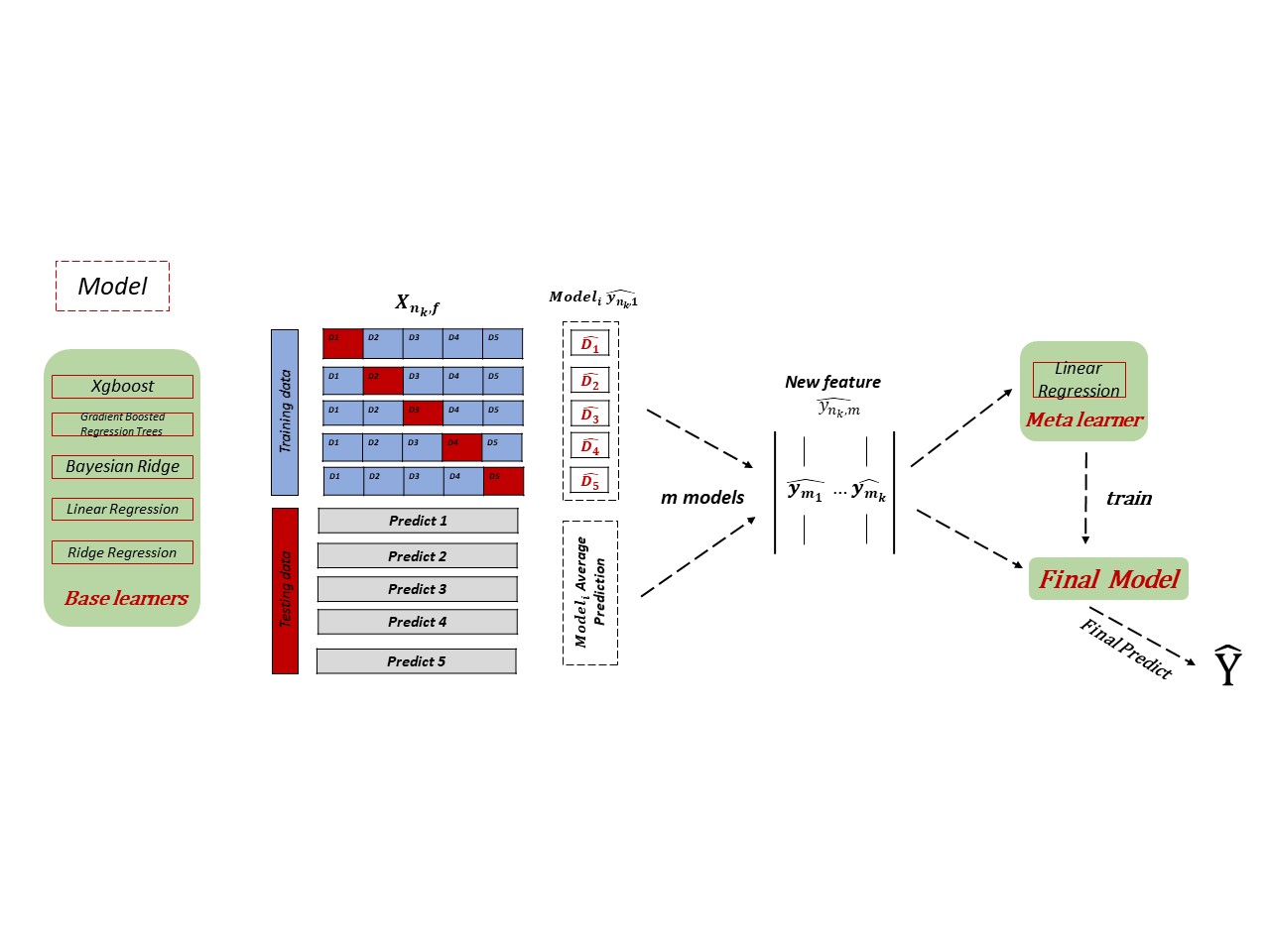
Scikit-learn implementation of stackimpute, a stacking approach for the imputation of multiple platforms methylation data. STACKIMPUTE has no requirement on the sequencing type of datasets and has already been proved that it has a good performance on Infinium Methylation 450K, Infinium MethylationEPIC (850K), Reduced representation bisulfite sequencing (RRBS), and Whole-Genome Bisulfite Sequencing (WGBS) data under different cases. It is able to share information between different samples. However, if you only have one sample, you could also use STACKIMPUTE to do the imputation. You would have a desirable result too. For a detailed description of the algorithm, please see our manuscript stackimpute: a universal stacking machine learning model for multi-platform methylation data imputation (2021).
stackimpute requires scikit-learn library. See the requirements.txt file for more details on required Python packages. We recommend you to create a new environment and install all required packages with:
pip install -r requirements.txt
The most convenient way to install stackimpute
pip install stackimpute
Besides, you could download this github repository,
git clone https://github.com/sixone11/stackimpute.git
and then install stackimpute by setup.py file
python setup.py install
- sort the bed file and filter the X,Y chromosomes
sort -k1,1V -k2,2n -k3,3n|grep -v chrX|grep -v chrY - the input file format (-i inputfile) without column names
- chromosome (with chr) ex: chr1
- the CpG sites' postions (1-based) ex: 1298332
- the methylation level of sample 1 (from 0 to 1) ex: 0.50
if you have more than 1 samples to impute then you could add more columns
- the methylation level of sample 2 (from 0 to 1)
- ....
- the methylation level of sample n (from 0 to 1)
you could do this by yourself or using our script to merge your original .bed file
python input_generation.py \
-i sample1.bed sample2.bed ..... \
-o mergefile.out
and the format of sample.bed should be chr"\t"postion start"\t"postion end"\t"methylation level (from 0 to 1)
- Optional: the feature file format (-f feature file) If you provide this file, you must have column names of it and the first 2 column names should be "chr" and "position".
-
chromosome (with chr)
-
the CpG sites' postions (1-based)
-
feature 1
-
feature 2
-
feature 3
-
feature 4
-
.....
- feature selection
You could first filter the features and leave the features that affect the model most and then use them to train our model. The top 50% feature was selected by default. If you want to modify the top feature rate, you could specify it by the parameter -p.
python random_feature_top_selection.py \-i mergefile.out (required) \-f feature.file (feature selction) \-p top feature rate (float:0~1, optional, default:0.5) \-o outdir(required)Parameter Description -i input file (required) -f feature file (required) -p top feature rate (float:0~1, optional, default:0.5) -m the species is mouse (optional) default: off -o outfile directory (required)
- Model training
python stackimpute.py -i mergefile.out(required) \
-f feature.file(optional) \
-t training rate(float:0~1, optianal) \
-s ( No need to follow other parameters after "-s", optional)
-o imputation.out(required)
| Parameter | Description |
|---|---|
| -i | input file (required) |
| -f | feature file (optional) |
| -t | training rate (float:0~1, optianal) if you don't specified this parameter, the whole dataset will be used to train the model. |
| -m | the species is mouse (optional) default: off |
| -s | If it is specified, the trained model will be saved. (optional, default: no) |
| -o | output file(required) |
- Load saved model
If you trained your model before and you want it to impute other samples, this part will help you achieve this goal.
python load_savemodel.py \
-i mergefile.out(required) \
-l model (required) \
-o outfile.out
| Parameter | Description |
|---|---|
| -i | input file (required) |
| -l | model file (required) |
| -o | output file(required) |
- Model evaluation
If you want to evaluate the performance of stackimpute, you could provide your input file and specify the missing rate which means we would use the total number of sites*missing rate as the number of sites used in validation set.
python stackimpute.py \
-i mergefile.out
-f feature.file (optional)\
-r missing rate (float:0~1, optional default:0.1) \
-o outfile
| Parameter | Description |
|---|---|
| -i | input file (required) |
| -f | feature file (optional) |
| -r | missing rate (float:0~1, optional, default:0.1) 这里check一下default |
| -m | the species is mouse (optional) default: off |
| -o | outfile(required) |
01 install stackimpute first
pip install stackimpute02 get the example data
datadir=$(python -c 'import importlib;print(importlib.util.find_spec("stackimpute.data").submodule_search_locations[0])' )03 evaluate the performance of stackimpute
This command will generate the evaluation of model from cross-validation. The metrics contains precision, recall, specificity, and sensitivity along with Area Under TPR-FPR Curve (AUC) and Matthews correlation coefficient (MCC). For the specific methylation level of each site, we adopted 2 mainly metrics to benchmark the performance of models, Root Mean Square Error (rmse) and the Pearson’s correlation coefficient (r) across individuals.
eval_stackimpute.py \
-i ${datadir}/input.tsv \
-r 0.1 \
-s \
-o ${TODAYOUT}/performance.o04 do the imputation
This command will generate 2 files. The imputation result (imputation.result) and the trained model (imputation.pkl).
impute.py \
-i ${datadir}/input_NA.tsv \
-t 0.2 \
-s \
-o imputation.result
The data/input.out was 450K microarray data of 3 samples from GSE62992.
The **data/input_NA.out ** was the same sample with data/input.out, but it has some NAs to simulate real input files.
If you find our research useful, please consider citing:
Please contact elya.liu at yiliu11@zju.edu.cn for questions.