

Implementation of GREAT in Python
You require Python 3.8 or newer installed on your system. In case you do not have
Python installed, we recommend installing Miniconda <https://docs.conda.io/en/latest/miniconda.html>_.
Options to install greatpy:
- Install the latest release of
greatpyfromPyPI <https://pypi.org/project/greatpy/>:
pip install greatpy- Install the latest development version:
pip install git+https://github.com/theislab/greatpy.git@main| Information | link |
|---|---|
| Create regulatory domains file (regdom) | notebook |
| Enrichment test (binomial/hypergeometric) | notebook |
| Plotting of results | notebook |
| Comparisons with GREAT | notebook |
Please refer to
greatpy is a bioinformatics method that associates custom genomic regions to Gene Ontology (GO) terms by weighting genomic neighborhoods. It is based on and inspired by and inspired by GREAT (Genomic Regions Enrichment of Annotations Tool).
GREAT figure issue from GREAT article
- Translate tab-separated files (
.tsvor.bedformat) containing the following information:- Transcription start site annotations:
chromosome_number \t position \t strand \t gene_name. - Chromosome sizes file should have the following columns :
chromosome_number \t chromosome_size.
- Transcription start site annotations:
See data for input files
regdom = greatpy.tl.create_regdom(
tss_file=Input_TSS_path, # eg : "../data/human/hg38/tss.bed"
chr_sizes_file=Input_chromosome_size_path, # eg : "../data/human/hg38/chr_size.bed"
association_rule="Basalplusextention",
out_path=path_save_output,
)Allowed association rules are:
BasalplusextentionOneClosetTwoCloset

- This step calculates the significance of a custom set of genomic annotations through peak-gene mapping, using distal cis-regulatory regions of the genome.
- Input files :
- test file should have the following columns :
chr \t chr_start \t chr_end. - regulatory domain file should have the following columns :
chr \t chr_start \t chr_end \t name \t tss strand - chromosome size file should have the following columns :
chromosome_number \t chromosome_size. - annotation file should have the following columns :
ensembl \t id \t name \t ontology.group \t gene.name \t symbol
See test cases for genomic input files.
res = greatpy.tl.enrichment(
test_file=Input_path_or_df, # eg : "../data/tests/test_data/input/10_MAX.bed"
regdom_file=regdom_path_or_df, # eg : "../data/human/hg38/regdom.bed"
chr_size_file=chromosome_size_path_or_df, # eg : "../data/human/hg38/chr_size.bed"
annotation_file=annotation_path_or_df, # eg : "../data/human/ontologies.csv"
)Allowed tests for this function such as :
binom(default True): it calculates the binomial p-value.hypergeom(default True): it calculates the hypergeometric p-value.
Additionally, it is also possible to apply a Bonferroni and/or FDR correction to the found p-values:
res = great.tl.set_fdr(res, alpha=0.05)
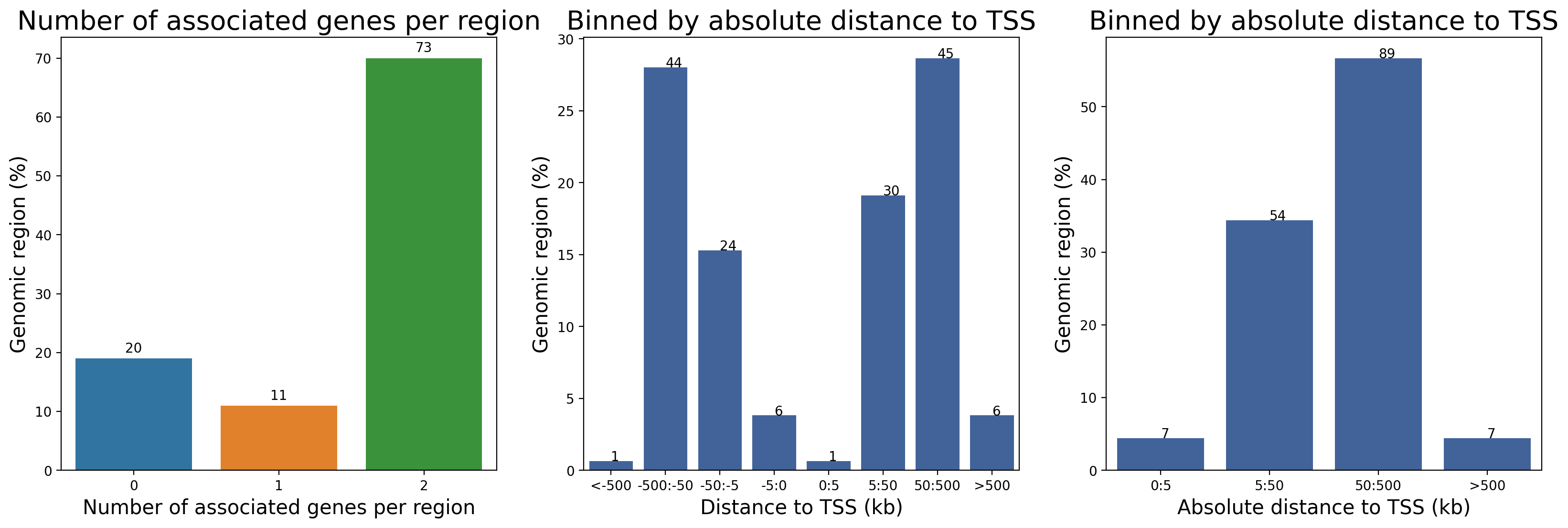
res = great.tl.set_bonferroni(res, alpha=0.05)- Number of genetic associations per genomic region.
- Distance to the associated gene TSS for each genomic region studied.
- Absolute distance to the associated gene TSS for each genomic region studied.
fig, ax = plt.subplots(1, 3, figsize=(30, 8))
greatpy.pl.graph_nb_asso_per_peaks(
Input_path_or_df, # eg : "../data/tests/test_data/input/10_MAX.bed"
regdom_path_or_df, # eg : "../data/human/hg38/regdom.bed"
ax[0],
)
greatpy.pl.graph_dist_tss(
Input_path_or_df, # eg : "../data/tests/test_data/input/10_MAX.bed"
regdom_path_or_df, # eg : "../data/human/hg38/regdom.bed"
ax[0],
)
greatpy.pl.graph_absolute_dist_tss(
Input_path_or_df, # eg : "../data/tests/test_data/input/10_MAX.bed"
regdom_path_or_df, # eg : "../data/human/hg38/regdom.bed"
ax[0],
)
plt.show()
plot = enrichment_df.rename(columns={"binom_p_value": "p_value", "go_term": "name"})
plt.figure(figsize=(10, 10))
great.pl.plot_enrich(plot)

test = ["name_bindome_biosample_1", "name_bindome_biosample_2", "..."]
tmp_df = great.tl.enrichment_multiple(
tests=test,
regdom_file="../data/human/hg38/regulatory_domain.bed",
chr_size_file="../data/human/hg38/chr_size.bed",
annotation_file="../data/human/ontologies.csv",
binom=True,
hypergeom=True,
)

Both binomial and hypergeometric tests may be susceptible to biases of which one must be aware to analyze the results critically. The binomial test reduces the hypergeometric bias by taking into account exactly the size of the regulatory domains of the genes, whereas the hypergeometric test compensates for the bias of the binomial test by counting each gene only once. The two types of tests are complementary and are recommended to be analyzed together.
See the changelog.
For questions and help requests, you can reach out in the scverse discourse. If you found a bug, please use the issue tracker.
If greatpy is useful for your research, please consider to cite as:
@software{greatpy,
author = {Ibarra, Mauger-Birocheau},
doi = {},
month = {},
title = {{greatpy}},
url = {https://github.com/theislab/greatpy},
year = {2022}
}@article{GREAT,
author = {McLean, C.
and Bristor, D.
and Hiller, M. et al.},
title = {GREAT improves functional interpretation of cis-regulatory regions},
journal = {Nat Biotechnol},
year = {2010},
month = {May},
day = {02},
volume = {28},
number = {495},
pages = {501},
doi = {10.1038/nbt.1630},
url = {https://doi.org/10.1038/nbt.1630}
}@Manual{rGREAT,
title = {rGREAT: GREAT Analysis - Functional Enrichment on Genomic Regions},
author = {Zuguang Gu},
year = {2022},
note = {https://github.com/jokergoo/rGREAT, http://great.stanford.edu/public/html/},
}