Manifiesto | Instrucciones de uso | Una recomendación final | Tabla de contenidos | Agradecimientos | Licencia
El propósito de este repositorio es acumular material didáctico que cualquier estudiante o investigador pueda usar para comenzar de manera autónoma a adquirir las habilidades necesarias para el trabajo, en colaboración o como miembro, en la UIBCDF. En ningún caso se puede entender este repositorio como una guía completa de cada uno de los temas que se presentan. Este repositorio es solamente un punto de partida.
Si eres una persona ajena a la UIBCDF y estás aquí por algún otro motivo, eres bienvenido a hacer uso de esta documentación y contribuir en su desarrollo. Esperamos que, si lo necesitas, interactues en el panel de problemas o questiones -Issues Board- del repositorio en GitHub sin ninguna reticencia.
Inicialmente el material se desarrollará en español para hacerlo más accesible dado el contexto de la UIBCDF y los potenciales usuarios. De esta manera facilitamos que cualquier estudiante, independientemente de sus conocimientos de inglés, se atreva a participar y contribuir de una manera activa.
El formato debe estar en la medida de lo posible basado en ficheros Markdown y Jupyter notebooks. Se requiere entonces, para la contribución al desarrollo de este repositorio, la instalación de ciertas herramientas en tu computadora, además de unos conocimientos mínimos para su uso:
- Mínimo conocimiento del sistema operativo de tu computadora (preferentemente Linux).
- Mínimo conocimiento de lo que es Git y GitHub.
- Mínimo conocimiento de Python.
- Instalación de un gestor de entornos de Python como Conda.
- Instalación de Jupyter lab o Jupyter notebook.
- Instalación de un mínimo de librerías científicas de Python.
Es por esto que, para que UIBCDF-Academia sea autoconsistente, el primer y segundo bloque de contenidos están dedicados al uso de UIBCDF-Academia y a la introducción del laboratorio computacional de investigación y sus herramientas -algunas de las mismas son necesarias para sacarle todo el provecho posible a este repositorio-. Una vez cubierta la exposición de los elementos generales que debemos conocer para trabajar, seguiremos con el material más específico de introducción a los conceptos físicos, químicos y biológicos para el trabajo de investigación en biología computacional y el diseño racional de moléculas con potencial farmacológico. Si perteneces a la UIBCDF o vas a colaborar con nosotros, como penúltimo bloque encontrarás la descripción de nuestro flujo de trabajo guíado por criterios de la Ciencia Abierta -OpenScience-. El último bloque es un glosario de las librerías científicas de utilidad en nuestros proyectos. En este caso, la función de estos notebooks no es pedagógica, sino documental. Allí puedes encontrar las referencias útiles para su instalación y uso, así como el enlace correspondiente al foro -en forma de cuestión abierta en el Issues Board del repositorio en GitHub- para preguntas y comentarios técnicos.
Finalmente, es pertinente añadir que para hacer que el desarrollo de este material sea más flexible, su estructura no será indexada de manera numerada.
El primer bloque de contenido de UIBCDF-Academia está dedicado a explicar [qué es UICDF-Academia][unidad:que_es], cómo se usa y cómo puedes contribuir a su desarrollo. Pero no es necesario que lleguemos allí para encontrar unas primeras pautas sobre como comenzar a usarlo. Brevemente, puedes hacer uso de este repositorio de tres maneras distintas descritas a continuación.
Si únicamente quieres leer el contenido de las unidades de UIBCDF-Academia, no debes hacer nada más que navegar de manera guiada por los ficheros de este repositorio mediante su web en GitHub. No necesitas conocimientos específicos sobre ningún aspecto técnico. Tu puerta de entrada a dichas unidades será el enlace que puedes encontrar en la sección Tabla de contenidos de este documento.
Existen unidades de UIBCDF-Academia en dos formatos: en formato Markdown -como es el caso de este documento- o en formato Jupyter notebook -un formato interactivo-. Si quieres ir más allá de simplemente acceder a la visualización de las unidades para su lectura, puedes, sin necesidad de instalar ni configurar nada, interaccionar con este repositorio mediante a un servidor remoto de Jupyter lab ofrecido por Binder. Debes saber que cualquier cambio que realices allí, no tendrá ningún impacto sobre este repositorio principal. Así que esta puede ser la manera perfecta para comenzar a jugar con las unidades de UIBCDF-Academia. Échale un ojo a la sección correspondiente de la unidad "Cómo se usa UIBCDF-Academia" o sigue al menos los tres siguientes pasos:
- Abre este enlace que nos lleva al servidor de mybinder de este repositorio o hacer
click en la insignia
en la parte superior de este documento. Y deja que se cargue.
- Haz click con el botón derecho sobre el archivo "README.md" -barra de archivos a la izquierda de la pantalla- y selecciona en el menú desplegado "Open With > Markdown Preview".
- Verás la copia de este documento en tu recien estrenado servidor remoto de Jupyter lab en Binder. Continua por allí su lectura, esa versión será a partir de ahora tu página principal de UIBCDF-Academia.
Eres atrevida o atrevido y quieres comenzar configurando tu propio entorno en el que interaccionar con tu copia local de UIBCDF-Academia. Enhorabuena por ese ímpetu. Te sugerimos entonces que saltes al contenido apropiado para ti en la sección extendida titulada "Cómo se usa UIBCDF-Academia" del primer bloque del contenido de este repositorioi. Allí encontrarás todo lo necesario para configurar tu entorno virtual, clonar UIBCDF-Academia e interaccionar con tu propia copia local.
Eres bienvenida o bienvenido a contribuir al desarrollo de este repositorio. Puede que quieras hacer correcciones, implementar nuevo contenido, o simplemente contribuir con sugerencias. Existe una sección en el primer bloque del contenido de UIBCDF-Academia donde puedes encontrar las indicaciones para hacerlo. Por favor, te invitamos a tener una participación activa. Desde el primer "Pull Request" que realices, aparecerá tu nombre en la lista de colaboradores a este proyecto.
La mejor actitud que debes tener para comenzar a ser un científico capaz de usar herramientas computacionales se resume perfectamente en la siguiente ilustración de xkcd (Randall Munroe):
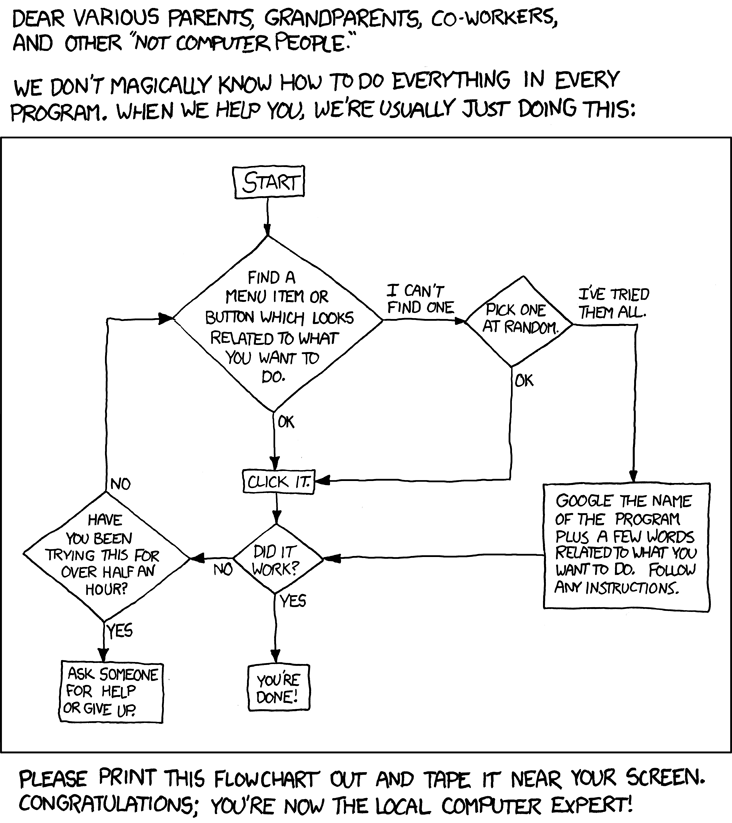
Al margen de la broma, perder el miedo, tener curiosidad y saber que gracias a internet es muy facil ser autodidacta, son los mejores consejos que te pueden dar. Te invitamos a jugar con los notebooks: ejecútalos, modifícalos, reprograma el código de sus celdas, añade más celdas para probar cosas nuevas. Comparte este material con quien tu quieras y siéntete en confianza para contribuir con dudas, problemas, sugerencias o soluciones en el panel de este repositorio en GitHub. Una vez alcanzado un nivel básico, es momento de estudiar los detalles con dedicación y profesionalidad. Pero eso ya es tarea tuya.
¡Ánimo!
Independientemente de cómo vayas a usar UIBCDF-Academia, puedes acceder desde ahora mismo a la primera tabla de contenidos de este repositorio. Recuerda, si lo estás haciendo desde la web principal del repositorio en GitHub únicamente podrás visualizar su contenido. Si lo estás haciendo desde Binder o desde tu propia copia local, podrás interaccionar con él.
Gracias a todos aquellos que de alguna manera ayudan a que este material crezca y sea útil. En especial a aquellos que por su colaboración activa pueden ser considerados autores o colaboradores.
Gracias también a los autores de la documentación y tutoriales citados en este repositorio. Gracias a los colegas que desarrollan las librerías de código abierto y software libre que aquí se usan. Y gracias también a xkcd (Randall Munroe) por hacer geniales ilustraciones sobre programación y ciencia, entre otras cosas, y permitir compartirlas libremente.
UIBCDF-Academia, cuya autoría corresonde a a los desarrolladores del repositorio uibcdf/Academia en GitHub, está registrado bajo licencia CC BY-NC-SA 4.0
Una copia del código legal de la licencia CC Attribution-NonCommercial-ShareAlike 4.0 International license's es incluida en este repositorio en inglés. El texto en español puede ser encontrado en este enlace..
UIBCDF-Academia by the contributors of the uibcdf/Academia GitHub repository is licensed under CC BY-NC-SA 4.0