Problem Setup
Larry Aagesen (@laagesen), David Montiel, Sourabh Kadambi (@sourabhkadambi), Sudipta Biswas (@SudiptaBiswas)
After the phase-field model formulation is developed, implemented in code, and verified, it can be set up to solve the scientific/engineering problem of interest. The purpose of this page is to give guidance on some of the important considerations when setting up your code to solve your specific problem, organized in the following sections:
Spatial dimension (1D vs. 2D vs. 3D)
In setting up a phase-field simulation, it is important to understand the role of spatial dimensions in the physics governing the problem, and how that might impact the simulation results and its interpretation. This is particularly important if simulations are being performed in reduced dimensions compared to the actual dimension of the problem, which is a common practice employed to reduce the computational costs.
It is likely that the physical behavior of a certain phenomenon might scale differently in 1D, 2D and 3D. This could be related to the physical processes in the bulk regions of the system or at the interfacial regions or both. For instance, the amount of interfacial region relative to the bulk phase region differs in 1D, 2D and 3D, and also governs many physical aspects of material behavior.
Therefore, it is important to determine how reduced dimensionality might affect predictions or comparison of simulation results with reality. In certain situations, on the other hand, it might be appropriate to setup the problem in reduced dimensions of 1D or 2D due to the inherent symmetry or directionality in the problem. Moreover, for simple microstructure geometries, it might be possible to setup the model in cylindrical or spherical coordinates, which can allow the simplicity of 1D computations while also capturing 2D and 3D behavior of the model accurately.
To demonstrate a case where simulation outcomes can significantly differ in different dimensions, we consider the single seed case of the homogeneous nucleation benchmark problem. A simple phase-field model with a single non-conserved order parameter describes an isothermal pure substance with one liquid phase (order parameter =
We consider a seed nucleus of
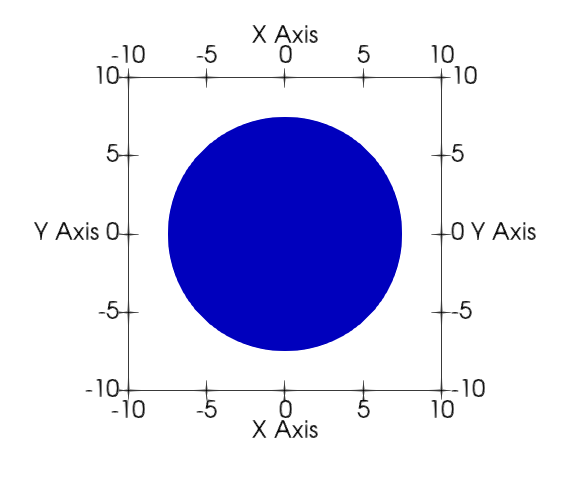
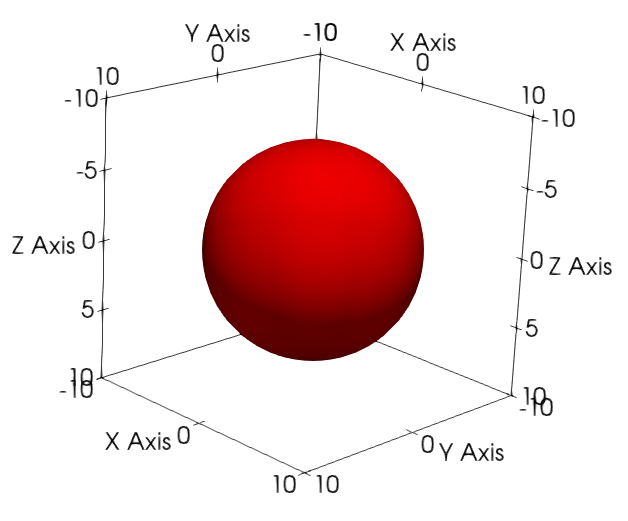
The time evolution of the order parameter, given by the Allen-Cahn equation, was solved using the MOOSE framework. Details of the numerical method can be found in Wu et al. (2021).
The simulations results of nucleus evolution are shown in the figures below: (left) order parameter profile measured radially from the center of domain at different evolution times; (right) radius as a function of evolution time. We see that the seed of same starting radius
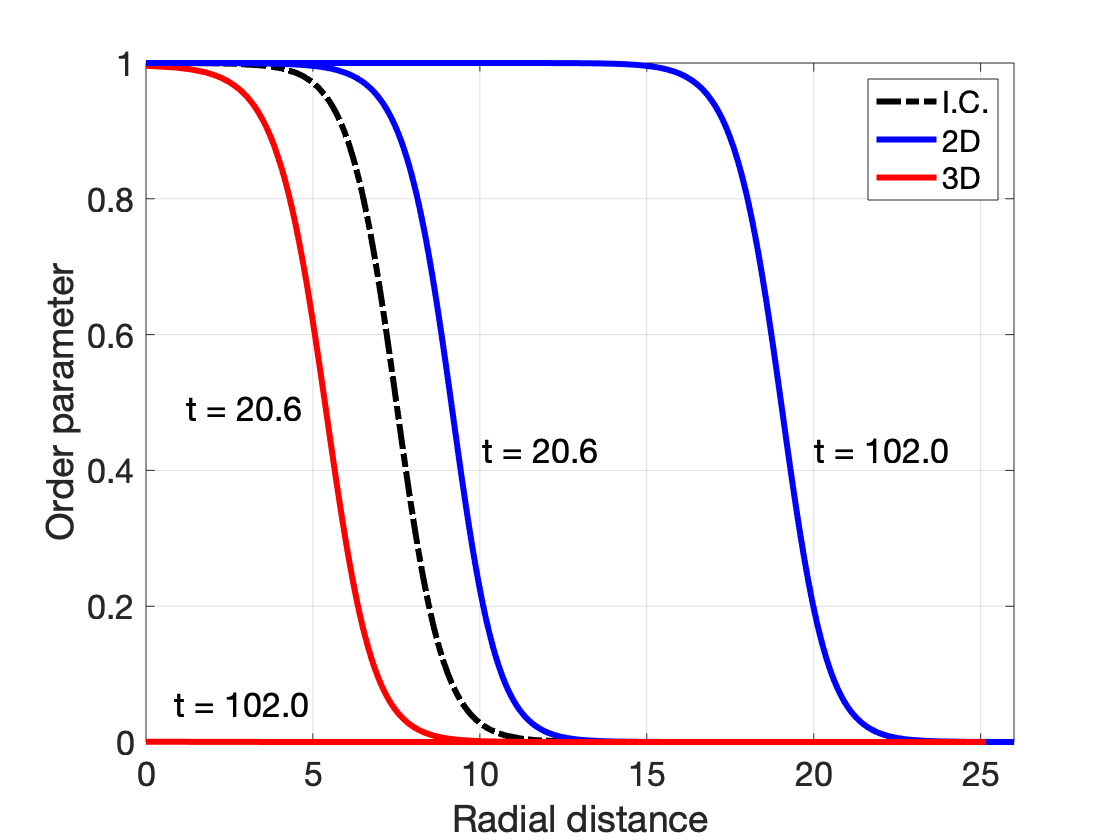
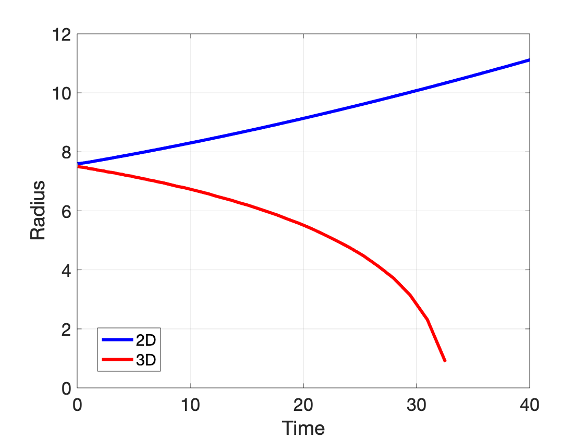
The role of dimensionality in homogeneous nucleation can be understood from the classical nucleation theory where the solid-liquid interface is modeled as a mathematically sharp interface. The interface is a 1D line in a 2D system and a 2D plane in a 3D system. The free energy of the nucleus particle
We can now apply the above sharp-interface analysis to our diffuse interface approximation in the phase-field model. For the given model parameters, we obtain
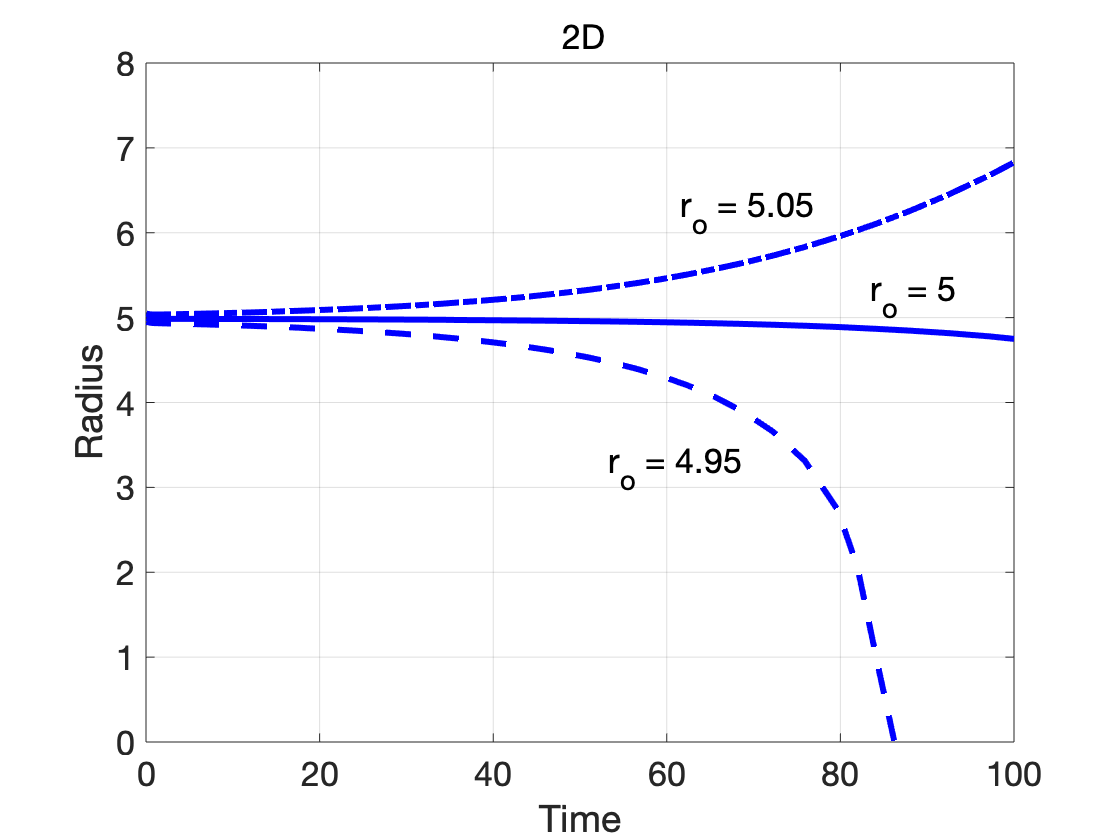
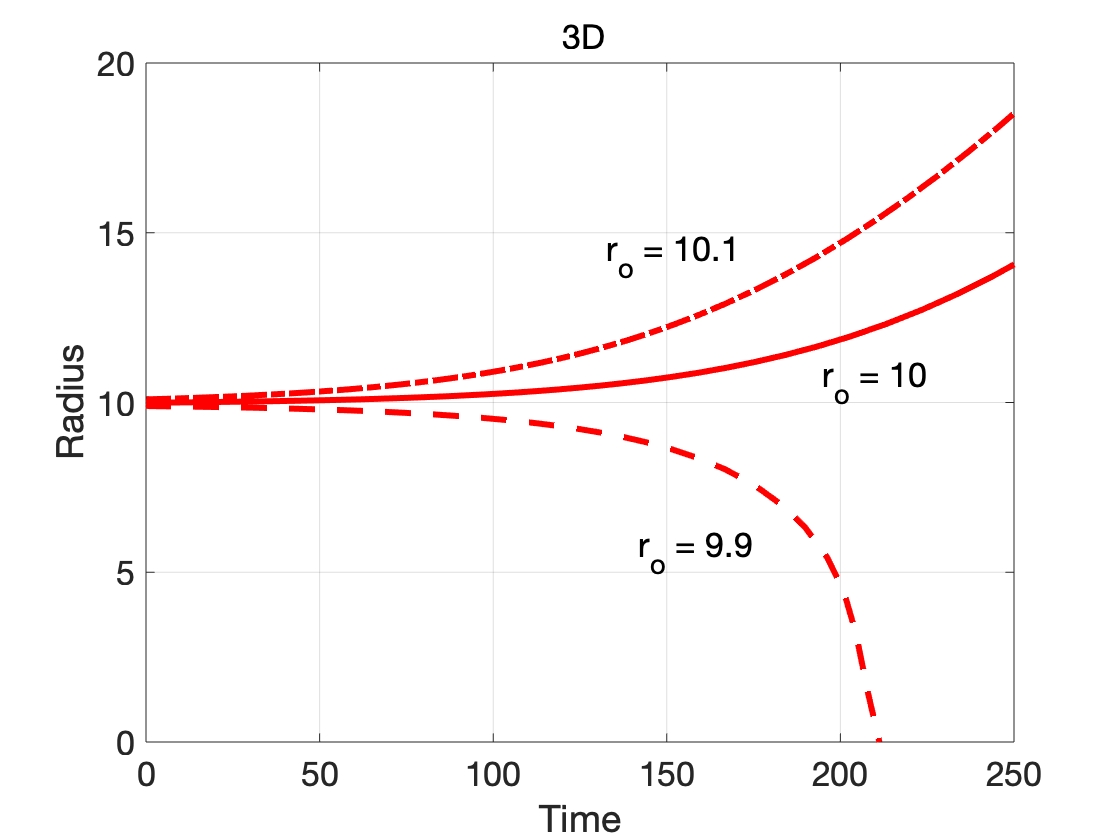
The dependence on dimensionality is further illustrated by considering cases where the initial radius is close to the critical radius:
Since the nucleus in the phase-field model is a diffuse-interface approximation of the classical sharp interface nucleus,
For simulations of the evolution of two or more phases, it is important to anticipate the expected equilibrium state of the system given the initial conditions. To do this, one needs the phase diagram as determined by the bulk contribution of the free energy density.
For example, in simulating phase separation following spinodal decomposition, it is common to define the initial state of the order parameter as a spatially uniform field with an added small 'noisy' perturbation with small amplitude. However, this order parameter must be within the spinodal region of the phase diagram, i.e., the region where spontaneous decomposition leads to a decrease in the free energy. Below we show two instances that demonstrate that choosing different values for the baseline order parameter,


Boundary conditions (BC) are required to solve for the governing equations in all phase field simulations. In general, every field must have a defined BC at every boundary of the system. The three most common types of boundary conditions (BC) for phase field simulation are:
- Dirichlet BC: The value of a field is specified at the boundary. This type of boundary condition is useful whenever we want to impose a value to an order parameter or field at one or more boundaries. Some common examples include setting a constant value for temperature to simulate a heat reservoir, or setting a constant value for a solid/liquid order parameter to indicate a fixed phase beyond the confines of the system.
- Neumann BC: The value of the spatial derivative of a field is specified at the boundary. This type of boundary conditions is useful to specify fluxes of fields at the boundary. For example, setting natural BC (a special case of Neumann BC) for a field at the boundary enforces that the normal component of gradient of that field is zero along that boundary. Therefore, if a flux for that field is proportional to this gradient, natural BC is equivalent to imposing zero flux at the boundary. This BC is convenient to ensure that the field is conserved. In addition, natural BCs are useful to exploit known symmetries in the morphology of domains: for example a spherical domain can be simulated using a quarter (in 2D) or eighth of a system (in 3D) by placing centering the sphere in a corner of the system and imposing natural BCs along the boundaries that define the corner.
- Periodic BCs: The value of the field in a boundary with periodic BC matches the value from the opposite boundary. These type of BCs are useful to simulate periodic domains, but also to minimize boundary effects. since the system does not interact with borders.
We used the results from Benchmark Problem 1a and Problem 1b to analyze the effect of boundary conditions. In addition, we solved for Cahn-Hilliard dynamics under the same initial condition and simulation parameters as problem 1 but using mixed boundary conditions, i.e., different boundary conditions for each boundary. We compare the results for each case at simulation time t = 1000. The simulations were carried out in the PRISMS-PF framework using a uniform square mesh with

As can be seen in the figure above, for periodic boundary boundaries the
Phase-field modeling is a diffuse interface approach, meaning that interfaces are represented by a smooth variation of one or more order parameters across the interface. The width of the interface is a function of the model parameters and the interface width is determined by model parameter choices. In some cases, an analytical expression is available that relates interface width to phase-field model parameters (such as free energy barrier height and gradient energy coefficient, which also impact the interfacial energy). In other cases, no analytical solution is available and the interface width must be approximated or determined numerically based on parameter choices. Such details are specific to the formulation being used.
Once the relationship between model parameters and interface width is understood, an appropriate selection of interface width needs to be made (while maintaining the correct interfacial energy for the system being studied). In some cases, the interface width in the phase-field model can be chosen to match the actual physical width of the interface being studied (such typically sub-nanometer). However, resolving a physically realistic interface width requires a sub-nanometer grid/mesh (grid/mesh convergence is described further in the next section). Using a physically realistic interface width often makes it computationally unfeasible to simulate systems large enough to be statistically representative. Therefore, in many cases an interfacial width that is much larger than the the physical width of an interface is used. In these cases, a careful balance between computational efficiency and model accuracy is needed; the interface width should be chosen to be small enough that the physics of the system are accurately represented, while maintaining adequate computational performance. A useful rule of thumb as a starting point is that the interface width should be at least an order of magnitude smaller than the smallest microstructural feature size of interest. Starting from this guideline, simulations of microstructural evolution with varying interface widths can be run to ensure that the choice of interface width does not affect the simulation results. A small test problem may be useful for testing convergence with respect to interface width; for example a shrinking circular grain embedded in another grain may be used for testing convergence of a grain growth model with respect to interface width, rather starting with large, costly simulations of hundreds of grains.
As an example of a mesh convergence study, we can consider Benchmark Problem 1 from the Phase-Field Community Hub. In this problem, which models spinodal decomposition using the Cahn-Hilliard equation, the width of the diffuse interface is 4.47, as defined by the Cahn-Hilliard equation and physical parameters in the problem statement. Given this interface width, we need to ensure there are a sufficient number of grid points (for finite difference schemes) or mesh elements (for finite element or finite volume schemes) through the diffuse interface to adequately resolve the variation of the composition order parameter.
In Problem 1b, a square domain with dimensions
The simulation initial conditions and the microstructures at
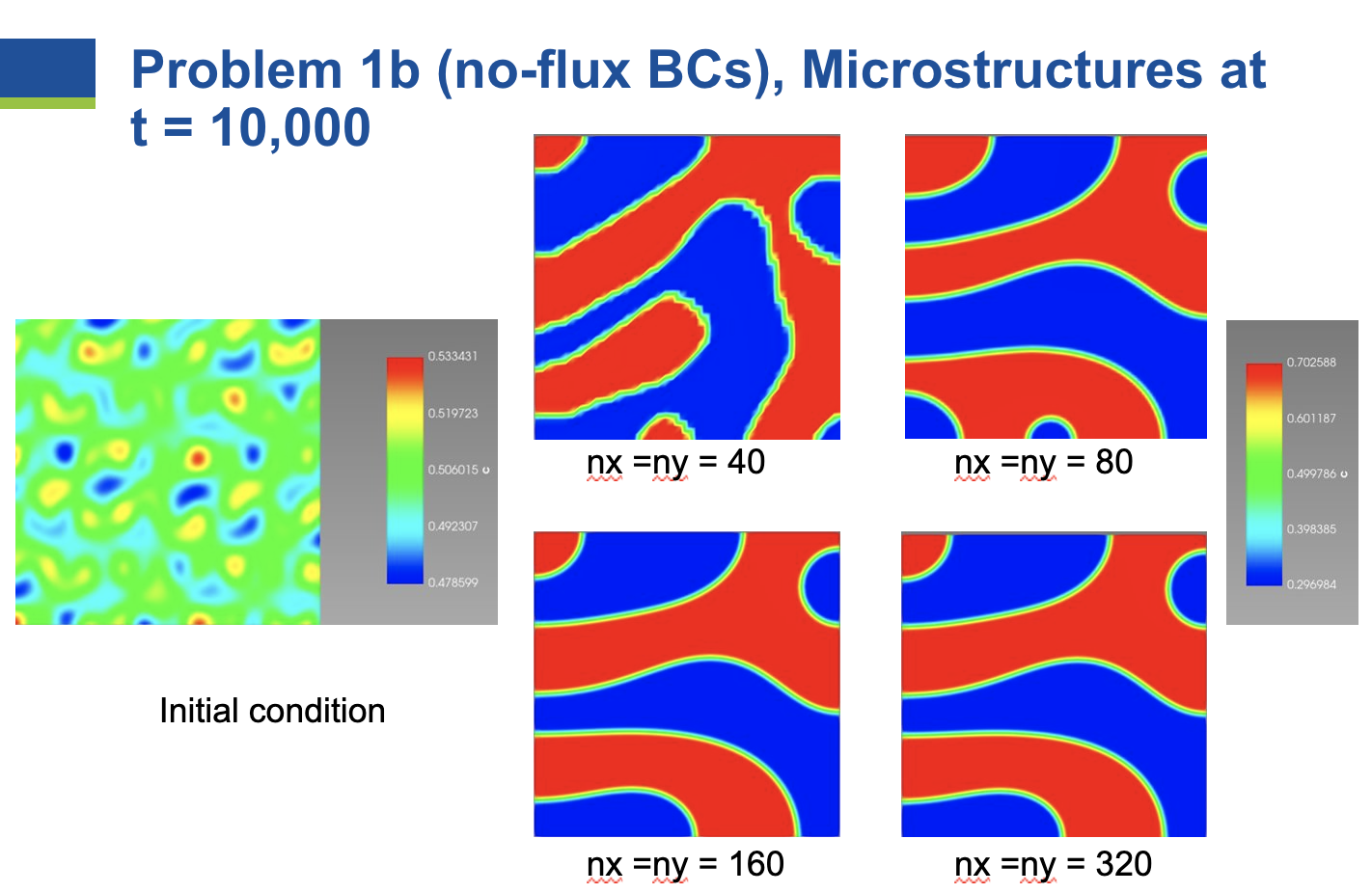
As the number of elements in each direction is increased from 40 to 80 to 160, changes in the microstructure are observed. However, once the number of elements increases to 320, no further changes are observed in the microstructure. Therefore, the problem is converged with respect to mesh resolution at
Carry out time step convergence studies. Try higher-order schemes, adaptive time stepping, etc. For explicit time integration, know your stability limit.
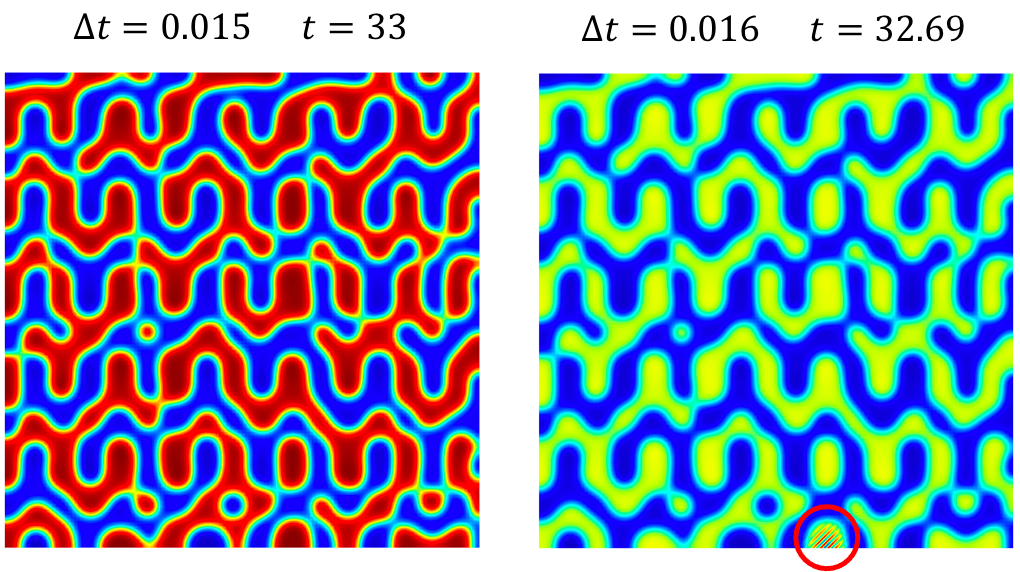
For codes that use explicit time integration, there is a maximum value of the time step beyond which the solution becomes numerically unstable. This stability limit can be determined by the Courant–Friedrichs–Lewy (CFL) condition and, in general, it strongly depends on the order of the spatial derivatives. Below we show the simulation results of Benchmark Problem 1b using time step values slightly below and slightly above the stability limit. We employed a forward Euler time-integration scheme and spatially discretized the system using
Codes that use implicit time stepping schemes may have fewer restrictions with respect to stability as time step size is increased, depending on the problem. However, discretization error still occurs in implicit schemes and increases with the size of the time step taken. Therefore, a convergence study should be carried out to ensure that the size of the time step does not affect the simulation results. As an example, we can again consider Benchmark Problem 1 from the Phase-Field Community Hub. Problem 1b was solved with the phase-field module from the MOOSE framework using

Adaptive time stepping can be useful to increase the time step size during time periods in the simulation where there are fewer microstructural changes, particularly for codes that use implicit time stepping schemes. However, convergence must still be checked for the parameters of the time stepping scheme being used. An example is the IterationAdaptiveDt time stepping scheme used in the MOOSE framework. This scheme attempts to increase or decrease the time step to keep the solver using a certain number of nonlinear iterations (controlled by the parameter optimal_iterations), within a window or plus or minus the parameter iteration_window. Higher values of optimal_iterations parameter result in higher time steps, and with that comes the risk of discretization error changing simulation results. As shown in the following, for Problem 1b, optimal_iterations values of 6 or 8 gave results consistent with the converged time step of 0.5, but when optimal_iterations was significantly increased to 15, changes in the microstructure resulted.

Oftentimes, the results of the phase field simulations are sensitive to the orientation and alignment of the key microstructural features with the mesh/grid points. To demonstrate this we pick a simple solidification problem with dendritic structure formation. In this case, we utilize the solidification example from the MOOSE-based phase field module. This example problem can quickly demonstrate dendritic structure formation including the formation of secondary dendritic arms in a computationally cost-effective manner. Here, we use 4-fold symmetry of the structure and vary the reference angles to misorient the dendritic arms with respect to the mesh. Dendritic structures corresponding to 0 and 45 degree reference angle is presented below:


It is noteworthy that the shape of the dendrite varies with orientation (as observed by the difference in the dendrite center). For better comparison, we rotate the

This highlights the slight differences between the dendrite shapes, especially the center and the secondary dendritic arms. Additionally, the growth rate of the solid also varies with orientation as observed by the change in solid area fraction over time:
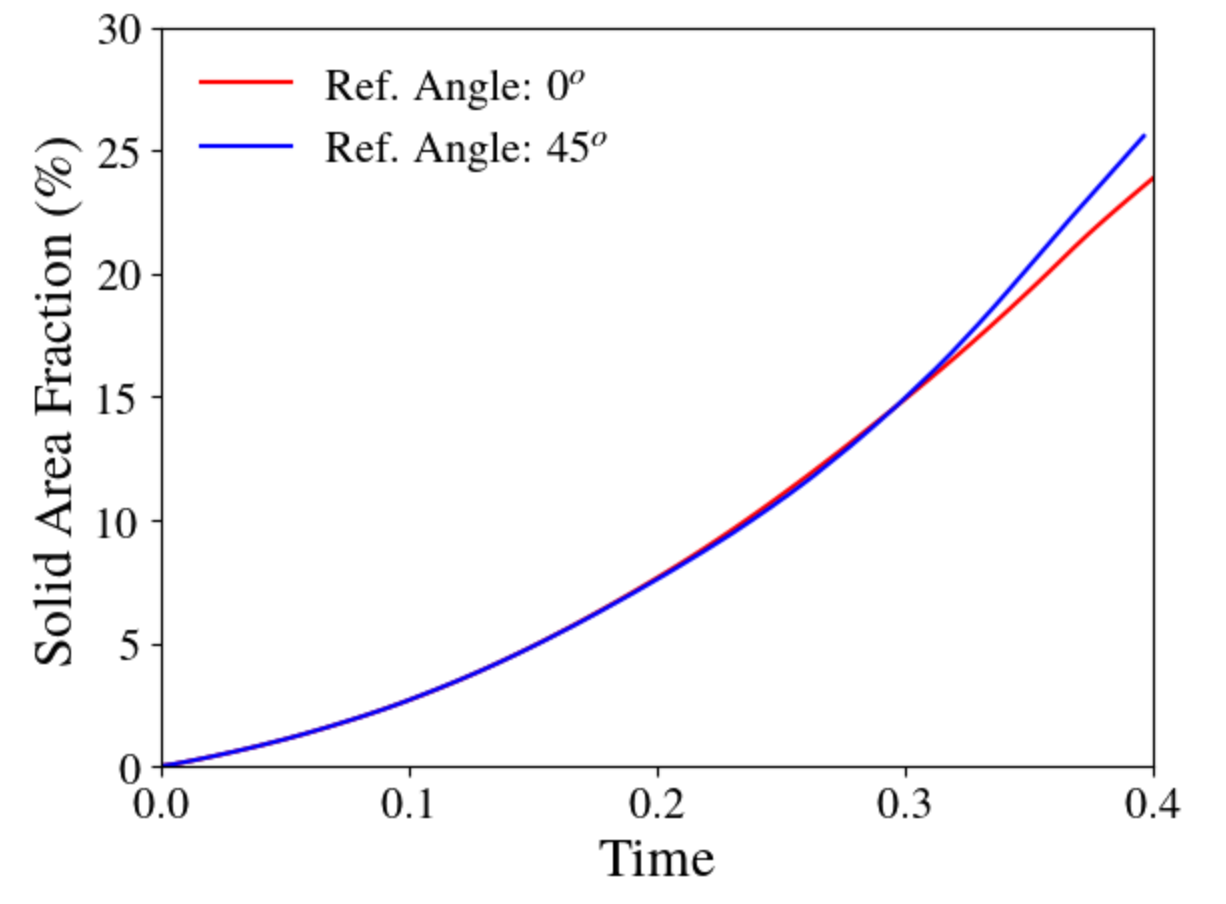
Thus, it is important to evaluate the effect of orientation on the results by misaligning the grids. Furthermore, these effects are influenced by the discretization of the mesh. Hence, it is important to ensure that the mesh is refined enough to properly resolve the interfaces (if necessary, run a mesh convergence study). For more information about strategies to simulate multiple dendrites with varying orientation, please refer to examples by Biswas et al. [1], Warren et al. [2], Dorr et al. [3], Ofori-Opoku et al. [4], and Pusztai et al. [5], among others.
The amount of time the simulation should be run depends on the science or engineering question to be answered, and on the system being studied. For many classic phase-field problems such as grain growth and coarsening, a characteristic feature size of the system increases with time, and the progress of microstructural evolution slows as the characteristic feature size increases. For example, in grain growth, at long times the mean grain diameter
It is also useful to be aware that the system may reach a metastable or stable equilibrium state with respect to system energy, in which case no further microstructural evolution will occur for increasing simulation time. In the grain growth example, if the system evolves to a single grain, stable equilbrium has been reached. If the system evolves to a two-grain configuration with a flat grain boundary between the grains, it has reached a metastable equilibrium; the system could still lower it energy by removing the grain boundary, but in that configuration, there is no kinetic driving force to remove it from the metastable state. To monitor for such possibilities, it is useful for the simulation to periodically output the total free energy of the system; a stable or metastable equilibrium state is indicated by a constant free energy with respect to time.
[1] Biswas et al., “Solidification and grain formation in alloys: a 2D application of the grand-potential-based phase-field approach”, Modelling and Simulation in Materials Science and Engineering, 30 (2022) 025013. https://doi.org/10.1088/1361-651X/ac46dc
[2] Warren et al., “Extending phase field models of solidification to polycrystalline materials”, Acta Materialia 51 (2003) 6035–6058. https://doi.org/10.1016/S1359-6454(03)00388-4
[3] Dorr et al., “A numerical algorithm for the solution of a phase-field model of polycrystalline materials”, Journal of Computational Physics 229 (2010) 626–641. https://doi.org/10.1016/j.jcp.2009.09.041
[4] Ofori-Opuku et al., “A quantitative multi-phase field model of polycrystalline alloy solidification”, Acta Materialia 58 (2010) 2155-2164. https://doi.org/10.1016/j.actamat.2009.12.001
[5] Pusztai et al., “Phase-field approach to polycrystalline solidification including heterogeneous and homogeneous nucleation”, Journal of Physics: Condensed Matter 20 (2008) 404205. https://doi.org/10.1088/0953-8984/20/40/404205