


conda install wgatools -c biocondagit clone https://github.com/wjwei-handsome/wgatools.git
cd wgatools
cargo build --releaseor just install from git:
cargo install --git https://github.com/wjwei-handsome/wgatools.gitA nix flake is also available. You can build from within the repo like this:
nix build .#wgatoolsOr directly install from github:
nix profile install github:wjwei-handsome/wgatoolsUsing nix, we can derive docker and singularity images:
nix build .#dockerImageFirst, we load the docker image into the local daemon:
docker load < resultIt's then possible to pack up a singularity image:
singularity build wgatools-$(git log -1 --format=%h --abbrev=8).sif docker-daemon://wgatools:latestThis can be useful when running on HPCs where it might be difficult to build wgatools.
> wgatools
wgatools -- a cross-platform and ultrafast toolkit for Whole Genome Alignment Files manipulation
Version: 0.1.0
Authors: Wenjie Wei <wjwei9908@gmail.com>
Usage: wgatools [OPTIONS] <COMMAND>
Commands:
maf2paf Convert MAF format to PAF format [aliases: m2p]
maf2chain Convert MAF format to Chain format [aliases: m2c]
paf2maf Convert PAF format to MAF format [aliases: p2m]
paf2chain Convert PAF format to Chain format [aliases: p2c]
chain2maf Convert Chain format to MAF format [aliases: c2m]
chain2paf Convert Chain format to PAF format [aliases: c2p]
maf-index Build index for MAF file [aliases: mi]
maf-ext Extract specific region from MAF file with index [aliases: me]
call Call Variants from MAF file [aliases: c]
tview View MAF file in terminal [aliases: tv]
stat Statistics for Alignment file [aliases: st]
dotplot TEST: Plot dotplot for Alignment file [aliases: dp]
filter Filter records for Alignment file [aliases: fl]
rename Rename MAF records with prefix [aliases: rn]
maf2sam TEST: maf2sam [aliases: m2s]
pafcov TEST: pafcov [aliases: pc]
pafpseudo TEST: generate pesudo maf from paf [aliases: pp]
chunk Chunk MAF file by length [aliases: ch]
help Print this message or the help of the given subcommand(s)
Options:
-h, --help Print help (see more with '--help')
-V, --version Print version
GLOBAL:
-o, --outfile <OUTFILE> Output file ("-" for stdout), file name ending in .gz/.bz2/.xz will be compressed automatically [default: -]
-r, --rewrite Bool, if rewrite output file [default: false]
-t, --threads <THREADS> Threads, default 1 [default: 1]
-v, --verbose... Logging level [-v: Info, -vv: Debug, -vvv: Trace, defalut: Warn]Each subcommand could be used with -h or --help to get more information.
wgatools gen-completion --shell fish > ~/.config/fish/completions/wgatools.fishReady to enjoy it!
Three mainstream formats(PAF, MAF, CHAIN) can be converted to each other.
For example, to convert MAF to PAF:
wgatools maf2paf test.maf > test.pafor to convert PAF to MAF:
wgatools paf2maf test.paf --target target.fa --query query.fa > test.mafTip
If you want to convert into MAF format, you should provide target and query genome sequence files in {.fa, .fa.gz}.
stdin and stdout are supported, so you can use pipes to chain commands together🪆:
cat test.maf | wgatools maf2paf | wgatools paf2maf -g target.fa -q query.fa > test.maf
wgatools paf2chain test.paf | wgatools chain2maf -g target.fa -q query.fa | wgatools maf2chain | wgatools chain2paf > funny.pafWe provide two modes for plot, for example:
- BaseLevel
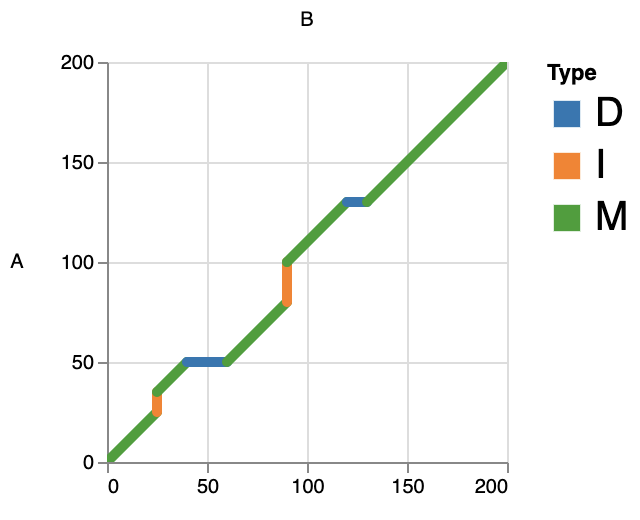
This mode can catch the alignment details in each record, such as matches, insertions and deletions. This can help us to better observe the local alignment.
wgatools dotplot -f paf test/testdotplot.paf > out.htmlBy default, INDELs smaller than 50bp are merged with adjacent match. You can also use the parameter -l, --length to specify the threshold.
In Interactive html, you can click on the legend to view only the types of interest, for example:
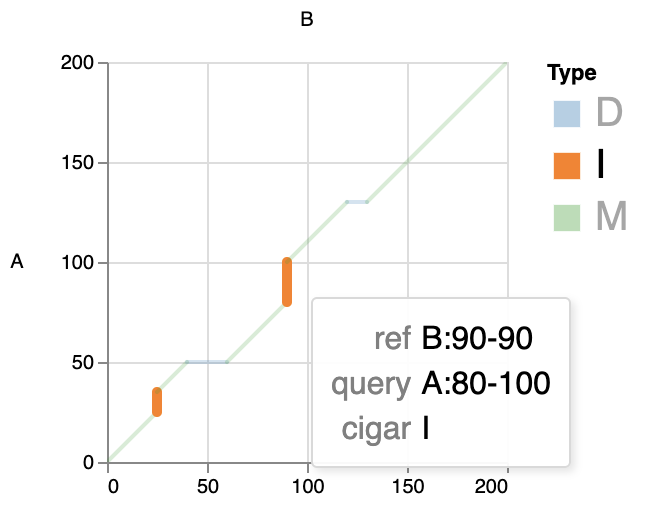
Warning
NOTE: For better interactivity, the zoom function is turned on. However, if there is too much data, the effect may be limited by your browser performance.
This simple example can be found in the test directory.
- Overview
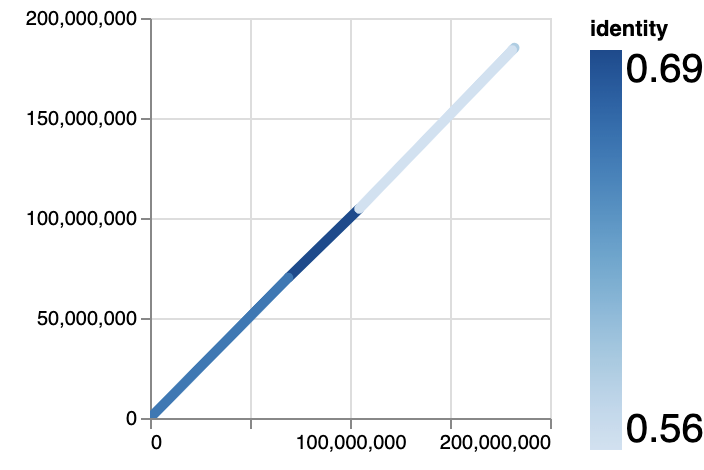
Similar to common dotplot scripts, it will draw each align record and color it according to identity.
wgatools dotplot test.maf -m overview > overview.html😎 For vega and DIY hackers, we also provide output in json(vega schema) and csv formats.
The line of MAF file is so long that it's hard to read. You can use maf-ext to extract specific region from MAF file with index:
wgatools maf-index test.maf
wgatools maf-extract test.maf -r chr1:1-10,chr2:66-888,chr3:100-50,chr_no:1-10,x:y-zTip
- Support multi-interval input, separated by commas
- Support
bedinput to specify interval - Mismatched interval are skipped and warned
View the MAF file in the terminal smoothly, and you can also specify the area to view:
wgatools tview test.maf
Press ◄► to slide left and right.
Press q to exit.
Press g to bring up the navigation window, where the left side is the optional sequence name, and the right side is the optional interval of the selected sequence, you can press Tab to switch the left and right selection windows, and you can press ▲▼ to select the sequence and interval
After input a legal interval, you can Press Enter to jump to the Destination. Or press Esc to exit the navigation window.
The MAF format completely records the alignment of each base, so it can be used to identify variants.
Supported explicit varaint types:
- SNP
- INS
- DEL
- INV
The default parameter does not output SNP and short INS and DEL (<50). The example is as follows:
wgatools call test/test.maf -s -l0Output vcf:
##fileformat=VCFv4.4
##INFO=<ID=SVLEN,Number=A,Type=Integer,Description="Length of structural variant">
##INFO=<ID=SVTYPE,Number=1,Type=String,Description="Type of structural variant">
##INFO=<ID=END,Number=1,Type=Integer,Description="End position of the longest variant described in this record">
##INFO=<ID=INV_NEST,Number=1,Type=String,Description="Varations nested within inversion">
##FORMAT=<ID=QI,Number=1,Type=String,Description="Query informations">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT sample
ref.chr8 181470034 . TG T . . SVTYPE=DEL;SVLEN=1;END=181470035 GT:QI 1|1:query.chr8@181989530@181989530@P
ref.chr8 181470279 . G C . . . GT 1|1
ref.chr8 181470292 . A G . . . GT 1|1
ref.chr8 181470431 . C G . . . GT 1|1
ref.chr8 181470609 . C A . . . GT 1|1
ref.chr8 181470641 . C T . . . GT 1|1
ref.chr8 181470774 . A AAACCAAGA . . SVTYPE=INS;SVLEN=8;END=181470774 GT:QI 1|1:query.chr8@181990269@181990277@P
ref.chr8 181470793 . G T . . . GT 1|1
ref.chr8 181470894 . C T . . . GT 1|1
ref.chr8 181470895 . A T . . . GT 1|1
ref.chr8 181470903 . G A . . . GT 1|1
Important
This function does not support the identification of chromosomal rearrangements such as DUP, as this requires the extraction of sequences for realignment.
You can split a huge MAF record into multiple records by length:
wgatools chunk -l 100 test/test.maf -o chunked.mafwgatools stat test.maf
wgatools stat -f paf test.paf
wgatools stat test.mafYou can filter some records by block length or query_size.
For example, to filter records that contig vs reference:
wgatools filter test.maf -q 1000000 > filt.mafFor all-to-all alignment paf file which produced by wfmash, you can filter some pairs by align-size:
wgatools filter all2all.paf -a 1000000 > filt.mafIn some practices, the chromosome name of ref and query are both called chr1, which is not easy to distinguish.
You can rename the sequence name in MAF file with a prefix:
wgatools rename --prefixs REF.,QUERY. input.maf > rename.mafIf you have alignment results for multiple genomes, you can use this command to calculate the alignment coverage on the genomes. It's optimized to use with wfmash output.
wgatools pafcov all.paf > all.cov.bedswgatools pafpseudo -f all.fa.gz all.paf -o out_dir -t 10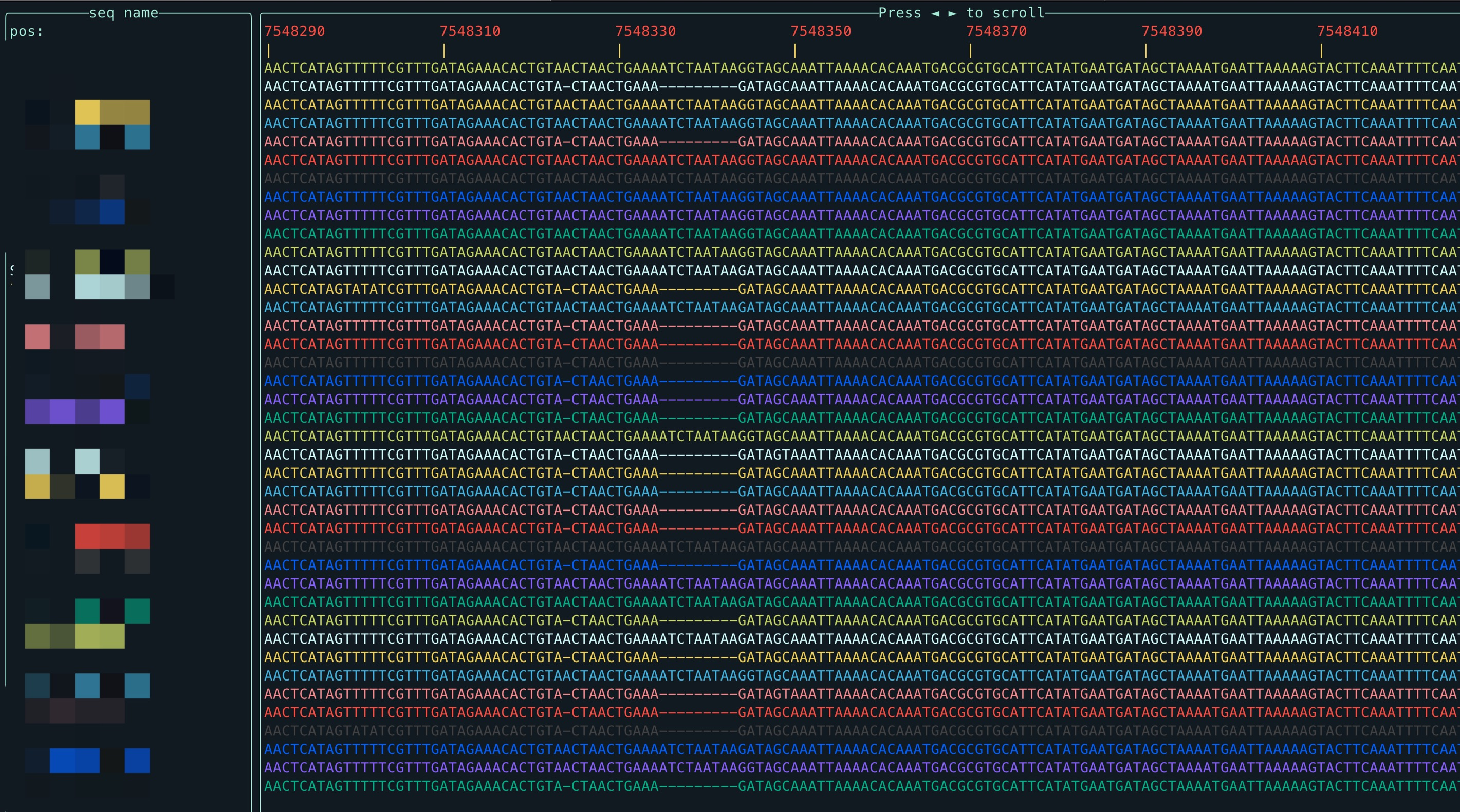
Some simple reader and iterator for PAF, MAF and Chain files:
use wgatools::parser::paf::PafReader;
use wgatools::parser::maf::MAFReader;
use wgatools::parser::chain::ChainReader;
fn main() {
let mut mafreader = MAFReader::from_path("test.maf").unwrap();
for record in mafreader.records() {
let record = record.unwrap();
println!("{:?}", record);
}
/// ...
}- Error detection and handling
- Test cases
- Documentations
- use
nomto parse CIGAR string - use
rayonto accelerate the speed of conversions - use
ratatuito visualize MAF file in terminal - ...
- SAM converter [really need?]
- Local improvement of alignment by re-alignment
- MAF -> GAF -> HAL
- output gvcf for variants
- call variants from PAF
Feel free to dive in! Open an issue or submit PRs.
MIT License © WenjieWei