
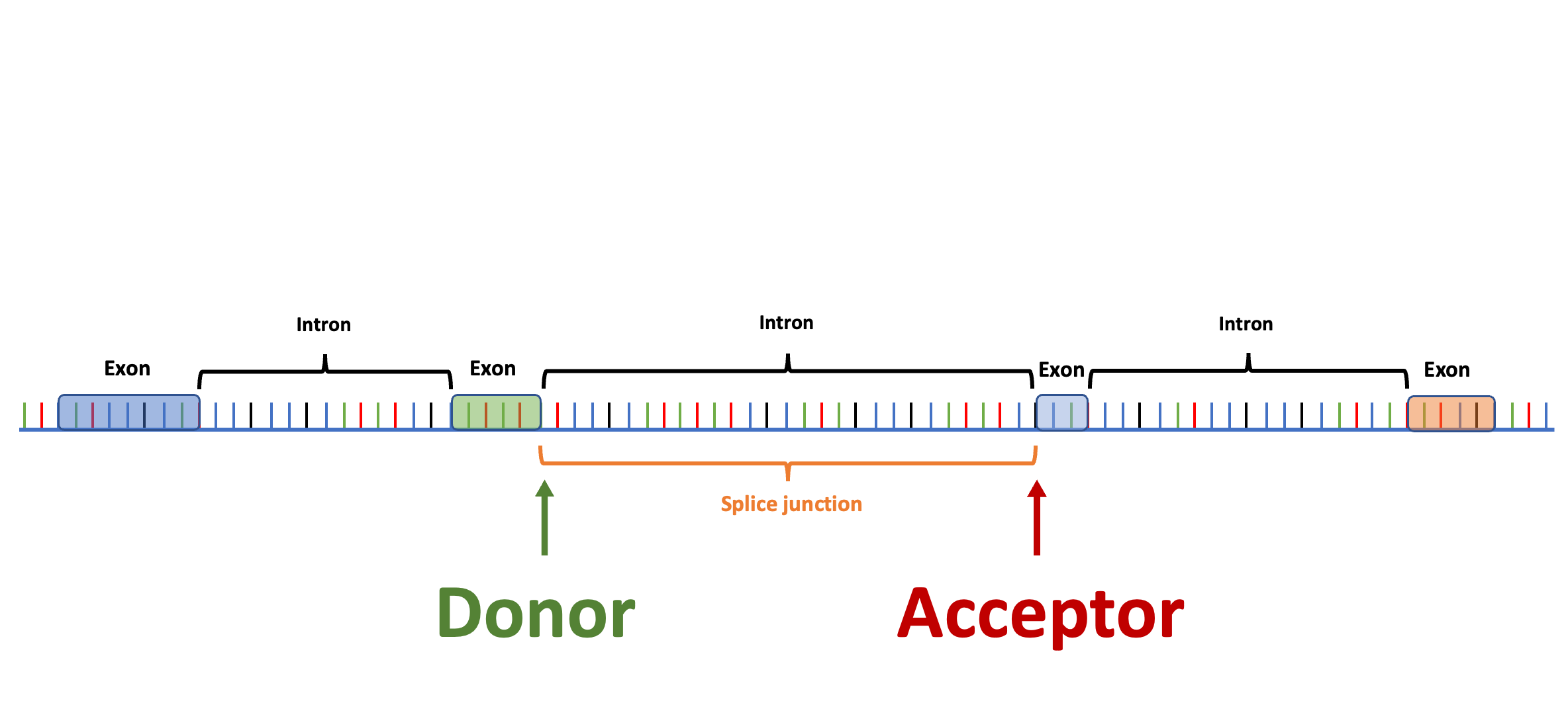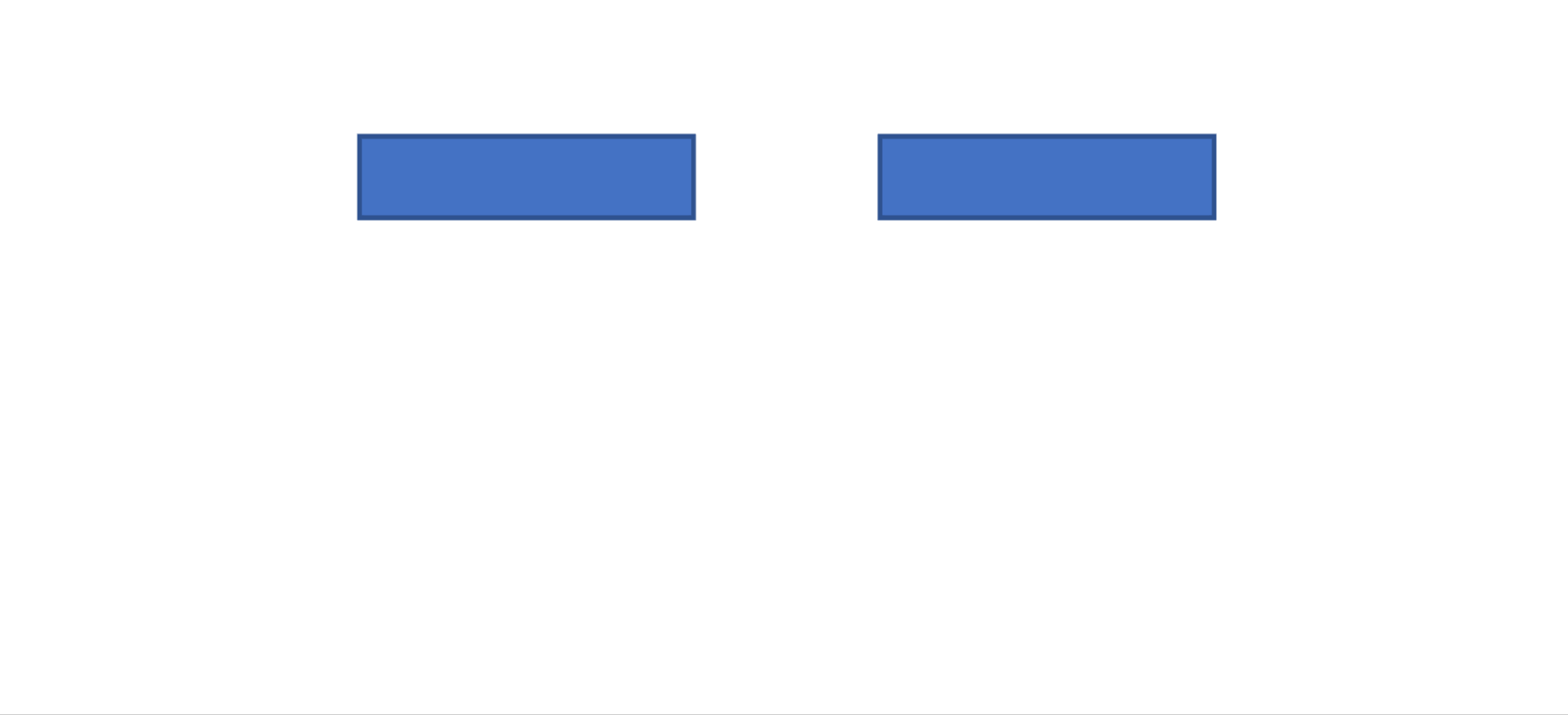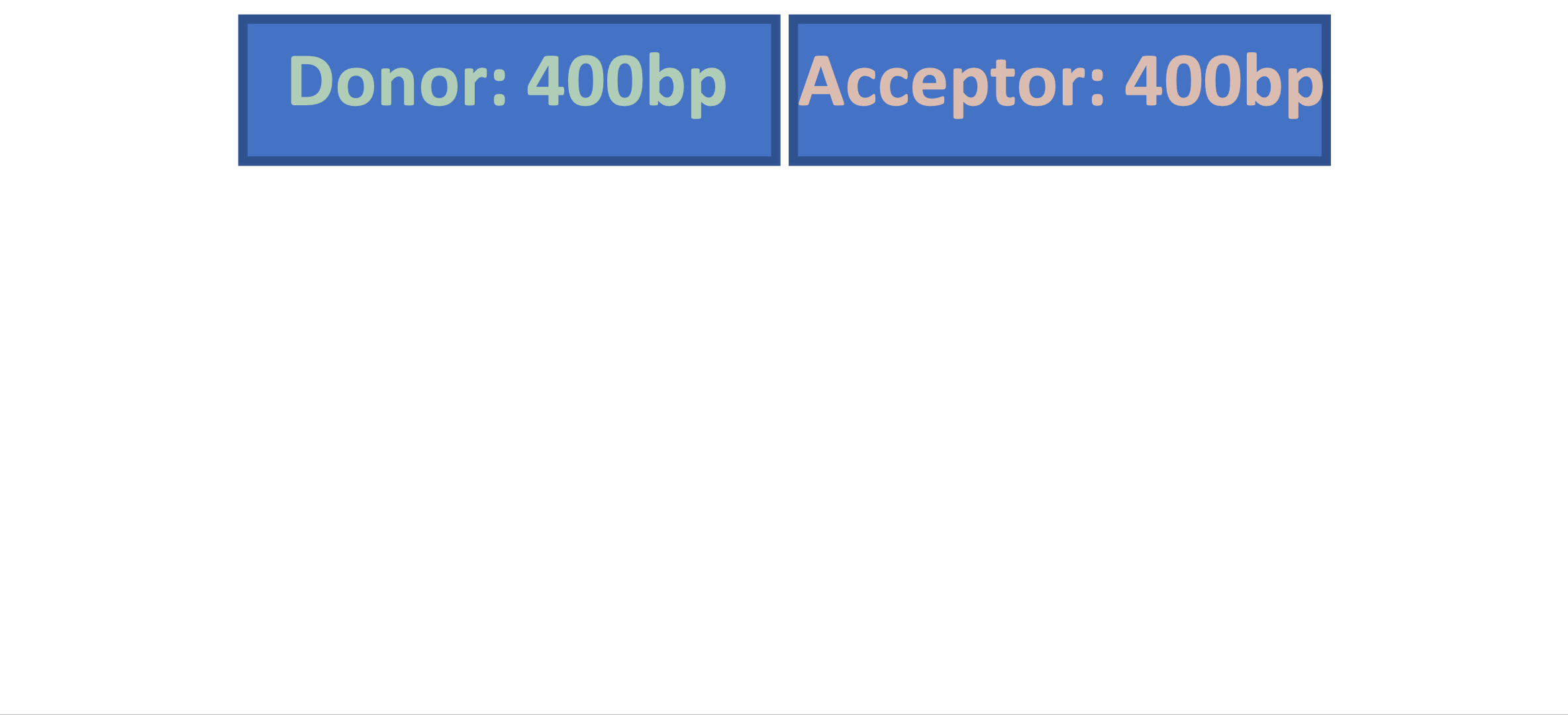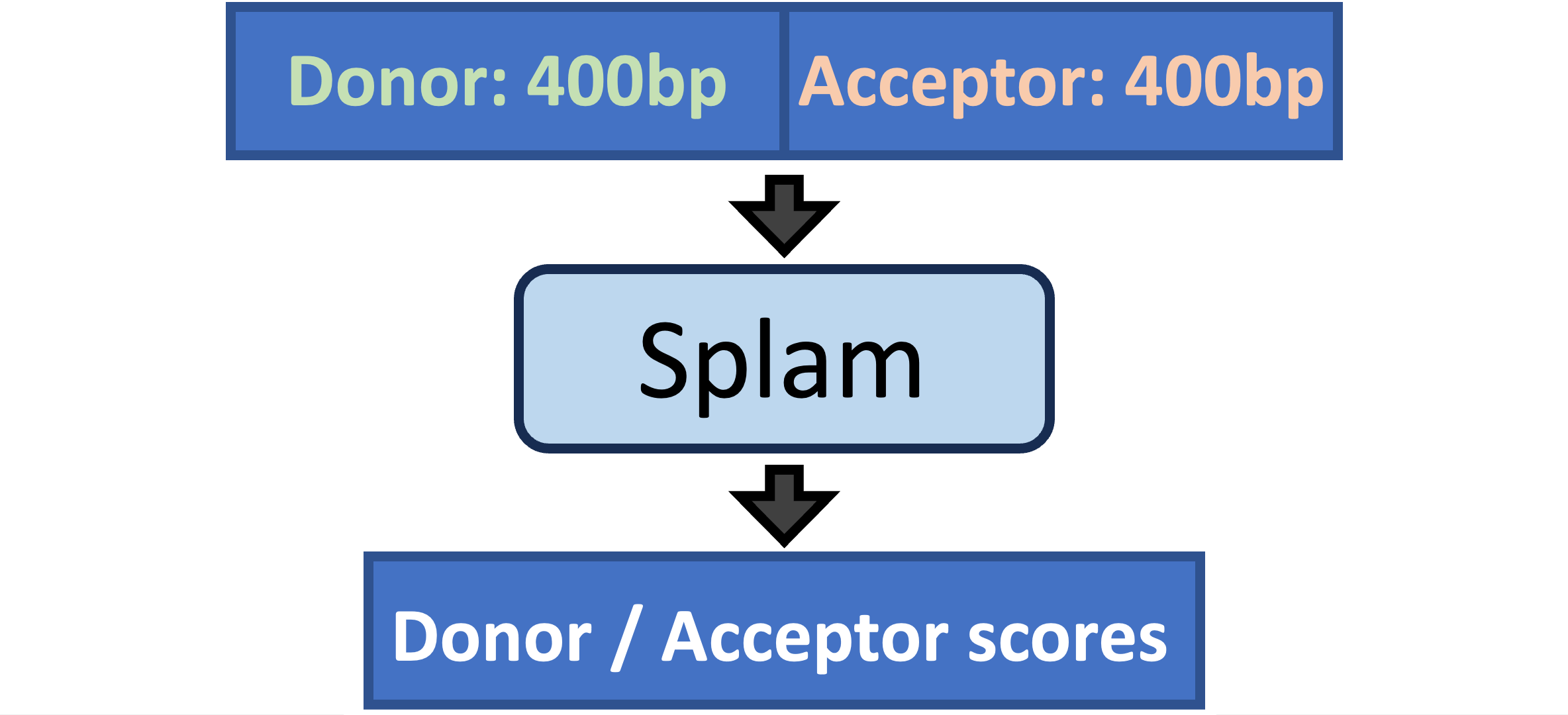
Splam is a splice site predictor utilizing a deep residual convolutional neural network for fast and accurate evaluation of splice junctions solely based on 400nt DNA sequences around donor and acceptor sites.
{kind=link}
{kind=link}
Why Splam❓#
- We need a tool to evaluate splice junctions & spliced alignments. Thousands of RNA-Seq datasets are generated every day, but there are no tools available for cleaning up spurious spliced alignments in these data. Splam addresses this problem!
- Splam-cleaned alignments lead to improved transcript assembly, which, in turn, may enhance all downstream RNA-Seq analyses, including transcript quantification, differential gene expression analysis, and more.
Who is it for❓#
If you are (1) doing RNA-Seq data analysis or (2) seeking a trustworthy way to evaluate splice junctions (introns), then Splam is the tool that you are looking for!
What does Splam do❓#
There are two main use case scenarios:
-
Improving your alignment file. Splam evaluates the quality of spliced alignments and removes those containing spurious splice junctions. This significantly enhances the quality of downstream transcriptome assemblies [Link].
-
Evaluating the quality of introns in your annotation file or assembled transcripts [Link].
Documentation#
📒 The full user manual is available here
Table of contents#
Installation#
Splam is on PyPi. This is the easiest installation approach. Check out all the releases here.
$ pip install splamYou can also install Splam from source
$ git clone https://github.com/Kuanhao-Chao/splam --recursive
$ cd splam/src/
$ python setup.py installQuick Start#
Running Splam is simple. It only requires three lines of code!
See these examples on Google Colab:
$ cd test
# Step 1: extract splice junctions in the alignment file
$ splam extract -P SRR1352129_chr9_sub.bam -o tmp_out_alignment
# Step 2: score all the extracted splice junctions
$ splam score -G chr9_subset.fa -m ../model/splam_script.pt -o tmp_out_alignment tmp_out_alignment/junction.bed
#Step 3: output a cleaned and sorted alignment file
$ splam clean -o tmp_out_alignment$ cd test
# Step 1: extract introns in the annotation
$ splam extract refseq_40_GRCh38.p14_chr_fixed.gff -o tmp_out_annotation
# Step 2: score introns in the annotation
$ splam score -G chr9_subset.fa -m ../model/splam_script.pt -o tmp_out_annotation tmp_out_annotation/junction.bed
#Step 3: output statistics of each transcript
$ splam clean -o tmp_out_annotation$ cd test
# Step 1: extract introns in the annotation
$ splam extract mouse_chr19.gff -o tmp_out_generalization
# Step 2: score introns in the annotation
$ splam score -G mouse_chr19.fa -m ../model/splam_script.pt -o tmp_out_generalization tmp_out_generalization/junction.bed
# Step 3: output statistics of each transcript
$ splam clean -o tmp_out_generalizationScripts for Splam model training & analysis#
All the scripts for Splam training and data analysis are in this GitHub repository.
Citation#
Kuan-Hao Chao*, Alan Mao, Steven L Salzberg, Mihaela Pertea*, "Splam: a deep-learning-based splice site predictor that improves spliced alignments ", bioRxiv 2023.07.27.550754, doi: https://doi.org/10.1101/2023.07.27.550754, 2023