Tree pattern matching user guide
The main principle of tree pattern matching is to define an evolutionary scenario, and search it as a subtree in a phylogenetic tree collection. This scenario is called tree pattern, and is built using a simple tool and click interface (the toolbox is in figure 1).
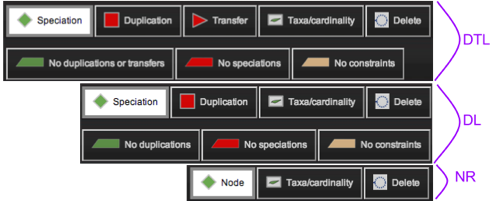
Figure 1 : Tool bar depends of the chosen databank. Tools are all available for DTL banks (reconciled with duplications, transfers and losses), and are minimum for not reconciled (NR) ones.
Every tool will be described throw examples, from the simplest pattern to more sophisticated ones. Some tools can have slightly different effects depending of which type is the explored databank (NR, DL or DTL).
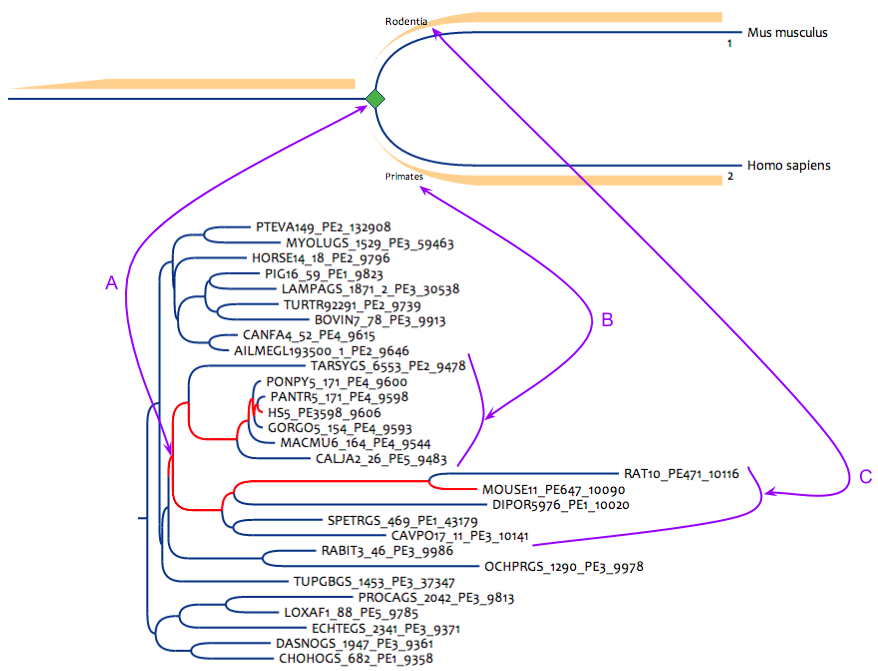
Figure 2 : Simple pattern to search mouse/human gene pairs from strict primates/rodents clades. This pattern can be built even for unreconciled tree databanks. (A) is the simple correspondance from the pattern node to the result node in the phylogenetic tree. (B) the mouse side must contain only rodent sequences. (C) the human side must contain only primate sequences.
-
Speciation/node tool: turns any leaf or node into a speciation or an untyped node for NR databanks.
-
Taxon/cardinality tool: permits to allow or forbid some taxonomic group in a leaf or in a part of the tree. If nothing is annotated, anything is allowed.
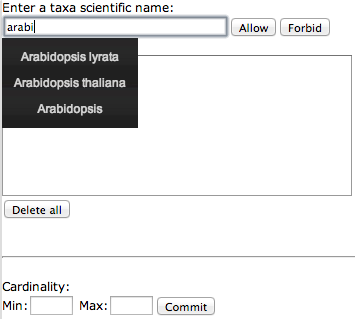
Figure 3: When using taxon/cardinality tools, a popup appears allowing to choose constraints about taxonomic representation at a leaf or in a part of the tree. It is also possible to control a range for the number of sequences represented under a specific node.
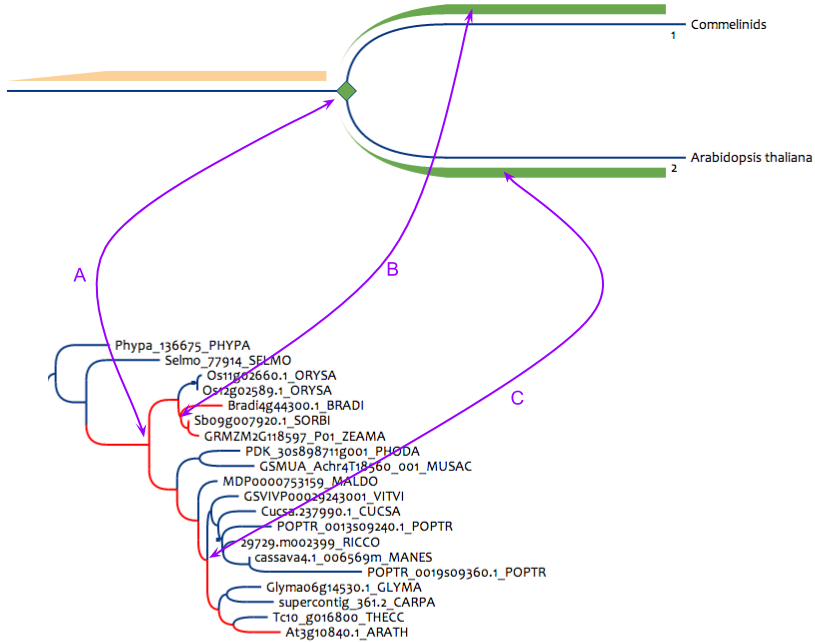
Figure 4 : Pattern to search super-orthologous gene pairs between Arabidopsis thaliana and any monocot. This pattern can be built only for reconciled databanks (at least with duplications, blues squares in the lower tree). (A) is the simple correspondance from the pattern node to the result node in the phylogenetic tree, this node must be a speciation. (B) the path between monocots and the speciation node must not contain duplications. (C) the path between Arabidopsis thaliana and the speciation node must not contain duplications.
-
no duplications or transfers tool: constraints the path in the phylogenetic tree, corresponding to the pattern branch, to contain only speciations.
-
no speciations tool: constraints the path in the phylogenetic tree, corresponding to the pattern branch, to contain only duplications or transfers.
-
no constraints tool: remove any constraints in the path in the phylogenetic tree.
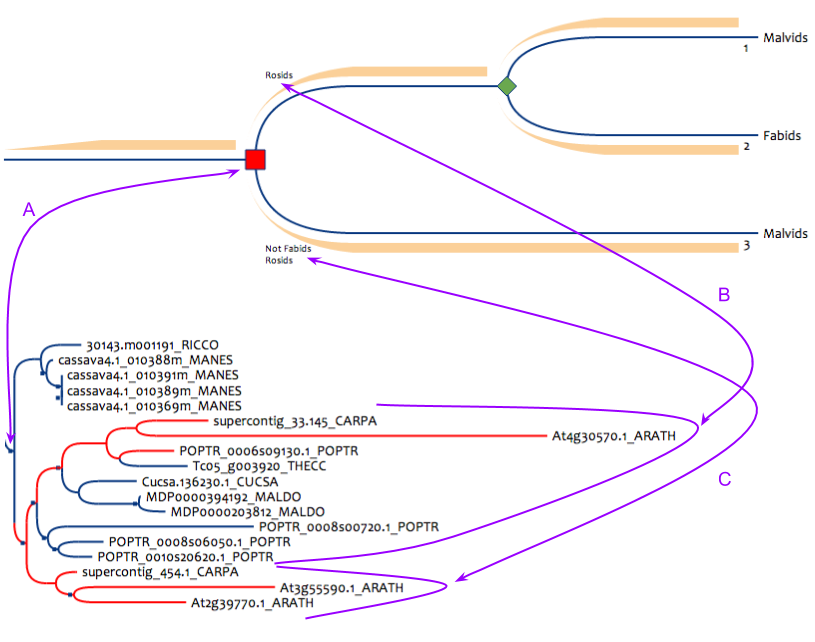
Figure 5 : Pattern to detect losses in fabids after a rosid specific duplication. This pattern can be built only for reconciled databanks (at least with duplications, blues squares in the lower tree). (A) is the correspondance from the pattern duplication node to the result duplication node in the phylogenetic tree, this node must be a speciation. (B) this subpart of the tree must contain only rosids. (C) this subpart of the tree must contain only rosids that are not fabids.
-
Duplication tool: Turns any leaf or node into a duplication node.
-
Transfert tool: Turns any leaf or node into a transfer node. Clicking several time on a transfer node allows to choose the sens of the transfer, or to let it undetermined. The databank must be reconciled using the DTL model.