Contact: Ruibang Luo
Email: rbluo@cs.hku.hk
Is called Clair. Clair is published in Nature Machine Intelligence. A preprint is available in bioRxiv
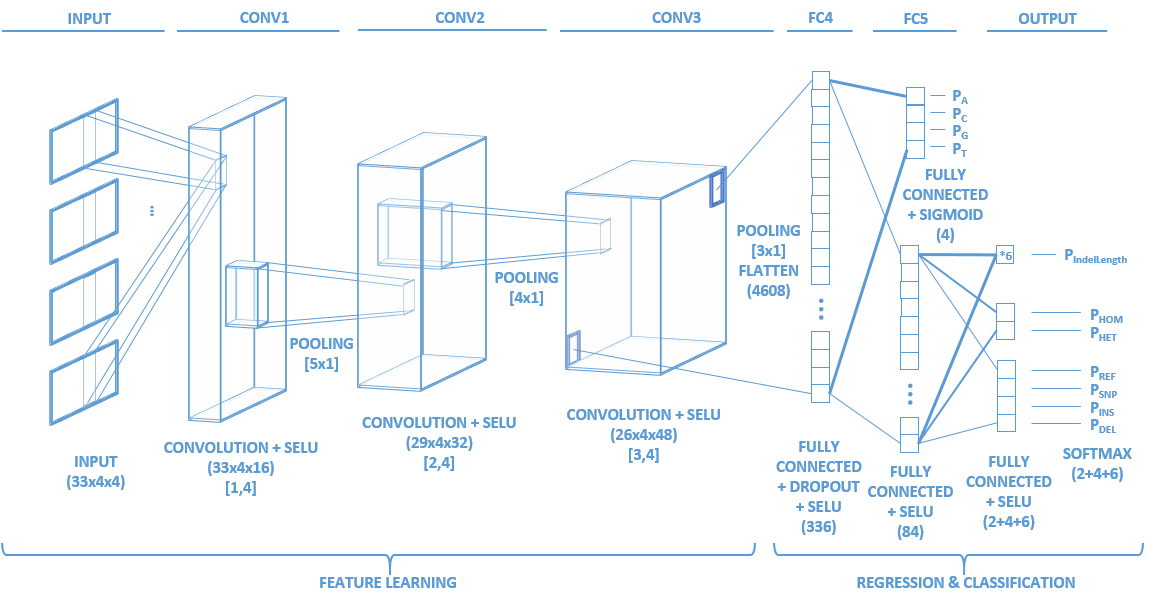
Identifying the variants of DNA sequences sensitively and accurately is an important but challenging task in the field of genomics. This task is particularly difficult when dealing with Single Molecule Sequencing, the error rate of which is still tens to hundreds of times higher than Next Generation Sequencing. With the increasing prevalence of Single Molecule Sequencing, an efficient variant caller will not only expedite basic research but also enable various downstream applications. To meet this demand, we developed Clairvoyante, a multi-task five-layer convolutional neural network model for predicting variant type, zygosity, alternative allele and Indel length. On NA12878, Clairvoyante achieved 99.73%, 97.68% and 95.36% accuracy on known variants, and achieved 98.65%, 92.57%, 87.26% F1 score on the whole genome, in Illumina, PacBio, and Oxford Nanopore data, respectively. Training Clairvoyante with a sample and call variant on another shows that Clairvoyante is sample agnostic and general for variant calling. A slim version of Clairvoyante with reduced model parameters produced a much lower F1, suggesting the full model's power in disentangling subtle details in read alignment. Clairvoyante is the first method for Single Molecule Sequencing to finish a whole genome variant calling in two hours on a 28 CPU-core machine, with top-tier accuracy and sensitivity. A toolset was developed to train, utilize and visualize the Clairvoyante model easily, and is publically available here is this repo.
Clairvoyante is published in Nature Communications
A PyTorch implementation of Clairvoyante is available at HKU-BAL/Clairvoyante-PyTroch.
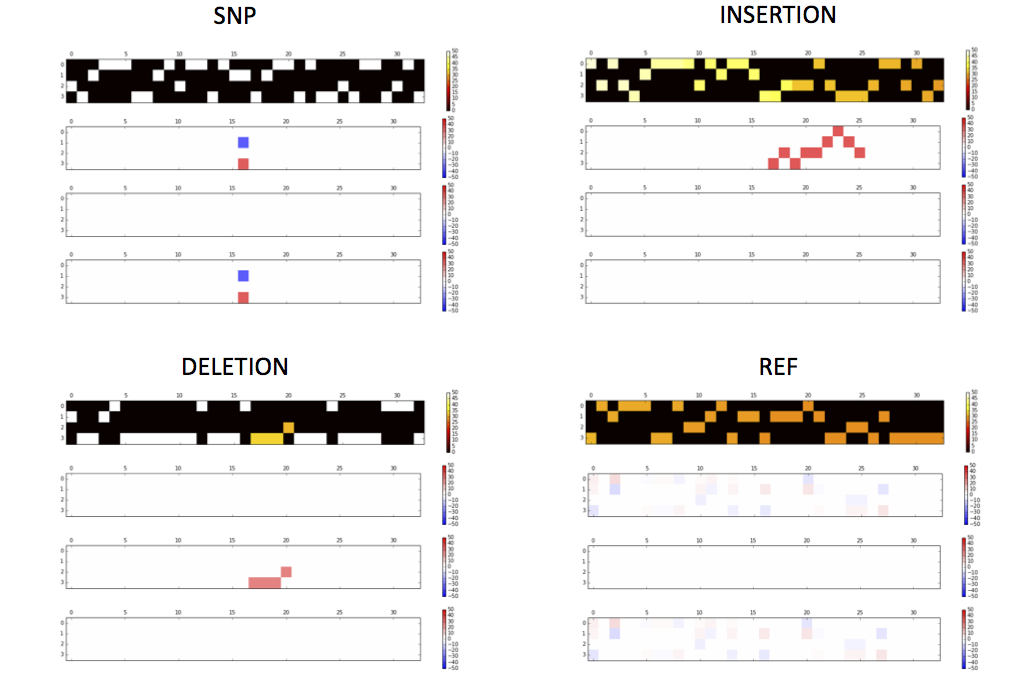
Activations of the conv1 hidden layer to a non-variant tensor

git clone --depth=1 https://github.com/aquaskyline/Clairvoyante.git
cd Clairvoyante
curl http://www.bio8.cs.hku.hk/trainedModels.tbz | tar -jxf -Make sure you have Tensorflow ≥ 1.0.0 installed, the following commands install the lastest CPU version of Tensorflow as well as other dependencies:
pip install tensorflow==1.9.0
pip install blosc
pip install intervaltree==2.1.0
pip install numpyTo check the version of Tensorflow you have installed:
python -c 'import tensorflow as tf; print(tf.__version__)'To do variant calling using trained models, CPU will suffice. Clairvoyante uses all available CPU cores by default in callVar.py, 4 threads by default in callVarBam.py. The number of threads to be used can be controlled using the parameter --threads. To train a new model, a high-end GPU and the GPU version of Tensorflow is needed. To install the GPU version of tensorflow:
pip install tensorflow-gpuThe installation of the blosc library might fail if your CPU doesn't support the AVX2 instruction set. Alternatively, you can compile and install from the latest source code available in GitHub with the "DISABLE_BLOSC_AVX2" environment variable set.
Clairvoyante was written in Python2 (tested on Python 2.7.10 in Linux and Python 2.7.13 in MacOS). For Python3.X user, run python port23.py which does a quickfix using 2to3 and inserts package namespace to sys.path to tackle python2-style import problem under python3.
Using pure Python interpreter on Clairvoyante is slow. Please refer to the Speed up with PyPy section for speed up.
conda create -n clairvoyante-conda-env -c bioconda clairvoyante
conda activate clairvoyante-conda-env
clairvoyante.pyThe conda environment has the Pypy intepreter installed, but doesn't have the required package intervaltree. The reason why the package is not installed by default is because it is not available in any conda repositories. To install the intervaltree for Pypy, after activation, please use the following commands:
wget https://bootstrap.pypa.io/get-pip.py
pypy get-pip.py
pypy -m pip install --no-cache-dir intervaltree==2.1.0
Download the models to a folder.
curl http://www.bio8.cs.hku.hk/trainedModels.tbz | tar -jxf -The commands above install the CPU version of TensorFlow in the virtual environment thus support only variant calling. To train a model, please install the GPU version of Tensorflow in the virtual environment:
conda remove tensorflow
conda install tensorflow-gpuUse conda deactivate to exit the virtual environment.
Use conda activate clairvoyante-conda-env to re-enter the virtual environment.
Without a change to the code, using PyPy python interpreter on some tensorflow independent modules such as dataPrepScripts/ExtractVariantCandidates.py and dataPrepScripts/CreateTensor.py gives a 5-10 times speed up. Pypy python interpreter can be installed by apt-get, yum, Homebrew, MacPorts, etc. If you have no root access to your system, the official website of Pypy provides a portable binary distribution for Linux. Following is a rundown extracted from Pypy's website (pypy-5.8 in this case) on how to install the binaries.
wget https://bitbucket.org/squeaky/portable-pypy/downloads/pypy-5.8-1-linux_x86_64-portable.tar.bz2
tar -jxf pypy-5.8-1-linux_x86_64-portable.tar.bz2
cd pypy-5.8-linux_x86_64-portable/bin
./pypy -m ensurepip
./pip install -U pip wheel intervaltree
# Use pypy as an inplace substitution of python to run the scripts in dataPrepScripts/Alternatively, if you can use apt-get or yum in your system, please install both pypy and pypy-dev packages. And then install the pip for pypy.
sudo apt-get install pypy pypy-dev
wget https://bootstrap.pypa.io/get-pip.py
sudo pypy get-pip.py
sudo pypy -m pip install intervaltree To guarantee a good user experience (good speed), pypy must be installed to run callVarBam.py (call variants from BAM), and callVarBamParallel.py that generate parallelizable commands to run callVarBam.py.
Tensorflow is optimized using Cython thus not compatible with pypy. For the list of scripts compatible to pypy, please refer to the Folder Stucture and Program Descriptions section.
Pypy is an awesome Python JIT intepreter, you can donate to the project.
You have a slow way and a quick way to get some demo variant calls. The slow way generates required files from VCF and BAM files. The fast way downloads the required files.
wget 'http://www.bio8.cs.hku.hk/testingData.tar'
tar -xf testingData.tar
cd dataPrepScripts
sh PrepDataBeforeDemo.shwget 'http://www.bio8.cs.hku.hk/training.tar'
tar -xf training.tarcd training
python ../clairvoyante/callVarBam.py \
--chkpnt_fn ../trainedModels/fullv3-illumina-novoalign-hg001+hg002-hg38/learningRate1e-3.epoch500 \
--bam_fn ../testingData/chr21/chr21.bam \
--ref_fn ../testingData/chr21/chr21.fa \
--bed_fn ../testingData/chr21/chr21.bed \
--call_fn chr21_calls.vcf \
--ctgName chr21
less chr21_calls.vcfcd training
python ../clairvoyante/callVar.py --chkpnt_fn ../trainedModels/fullv3-illumina-novoalign-hg001+hg002-hg38/learningRate1e-3.epoch500 --tensor_fn tensor_can_chr21 --call_fn tensor_can_chr21.vcf
less tensor_can_chr21.vcf| Input | File name |
|---|---|
| BAM | GIAB_v3.2.2_Illumina_50x_GRCh38_HG001.bam |
| Reference Genome | GRCh38_full_analysis_set_plus_decoy_hla.fa |
| BED for where to call variants | GRCh38_high_confidence_interval.bed |
- If no BED file was provided, Clairvoyante will call variants on the whole genome
python clairvoyante/callVarBamParallel.py \
--chkpnt_fn trainedModels/fullv3-illumina-novoalign-hg001+hg002-hg38/learningRate1e-3.epoch500 \
--ref_fn GRCh38_full_analysis_set_plus_decoy_hla.fa \
--bam_fn GIAB_v3.2.2_Illumina_50x_GRCh38_HG001.bam \
--bed_fn GRCh38_high_confidence_interval.bed \
--sampleName HG001 \
--output_prefix hg001 \
--threshold 0.125 \
--minCoverage 4 \
--tensorflowThreads 4 \
> commands.sh
export CUDA_VISIBLE_DEVICES=""
cat commands.sh | parallel -j4
vcfcat hg001*.vcf | vcfstreamsort | bgziptabix hg001.vcf.gzparallel -j4will run 4 commands in parallel. Each command using at most--tensorflowThreads 4threads.vcfcat,vcfstreamsortandbgziptabixare a part of vcflib.- If you don't have
parallelinstalled on your computer, tryawk '{print "\""$0"\""}' commands.sh | xargs -P4 -L1 sh -c. CUDA_VISIBLE_DEVICES=""makes GPUs invisible to Clairvoyante so it will use CPU only. Please notice that unless you want to runcommands.shin serial, you cannot use GPU because one running copy of Clairvoyante will occupy all available memory of a GPU. While the bottleneck ofcallVarBam.pyis at the CPU onlyCreateTensor.pyscript, the effect of GPU accelerate is insignificant (roughly about 15% faster). But if you have multiple GPU cards in your system, and you want to utilize them in variant calling, you may want split thecommands.shin to parts, and run the parts by firstlyexport CUDA_VISIBLE_DEVICES="$i", where$iis an integer from 0 identifying the seqeunce of the GPU to be used.
clairvoyante/callVar.py outputs variants in VCF format with version 4.1 specifications.
Clairvoyante can predict the exact length of insertions and deletions shorter than or equal to 4bp. For insertions and deletions with a length between 5bp to 15bp, callVar guesses the length from input tensors. The indels with guessed length are denoted with a LENGUESS info tag. Although the guessed indel length might be incorrect, users can still benchmark Clairvoyante's sensitivity by matching the indel positions to other callsets. For indels longer than 15bp, callVar.py outputs them as SV without providing an alternative allele. To fit into a different usage scenario, Clairvoyante allows users to extend its model easily to support exact length prediction on longer indels by adding categories to the model output. However, this requires additional training data on the new categories. Users can also increase the length limit from where an indel is outputted as a SV by increasing the parameter flankingBaseNum from 16bp to a higher value. This extends the flanking bases to be considered with a candidate variant.
wget 'http://www.bio8.cs.hku.hk/testingData.tar'
tar -xf testingData.tar
cd clairvoyante
python demoRun.py
Please visit: jupyter_nb/demo.ipynb
Interactively visualizing Input Tensors, Activation in Hidden Layers and Comparing Predicted Results (Suggested)
jupyter_nb/visualization.ipynb
getTensorAndLayerPNG.py. You will need to input a model, a file with one or more tensors created by CreateTensor.py and optionally a file with the truth variants created by GetTruth.py. The script will create 7 PNG figures for each line of tensor, including 1) input tensors; 2) conv1 layer activations; 3) conv2 layer activations; 4) conv3 layer activations; 5) fc4 layer activations; 6) fc5 layer activations and 7) output predictions and the truth.
The --olog_dir option provided in the training scripts outputs a folder of log files readable by the Tensorboard. It can be used to visualize the dynamics of parameters during training at each epoch.
You can also use the PCA and t-SNE algorithms provided by TensorBoard in the Embedding page to analyze the predictions made by a model. clairvoyante/getEmbedding.py helps you to prepare a folder for the purpose.
You can also run the program to get the parameter details.
dataPrepScripts/ |
Data Preparation Scripts. Outputs are gzipped unless using standard output. Scripts in this folder are compatible with pypy. |
|---|---|
ExtractVariantCandidates.py |
Extract the position of variant candidates. Input: BAM; Reference FASTA. Important options: --threshold "Minimum alternative allelic fraction to report a candidate"; --minCoverage "Minimum coverage to report a candidate". |
GetTruth.py |
Extract the variants from a truth VCF. Input: VCF. |
CreateTensor.py |
Create tensors for candidates or truth variants. Input: A candidate list; BAM; Reference FASTA. Important option: --considerleftedge "Count the left-most base-pairs of a read for coverage even if the starting position of a read is after the starting position of a tensor. Enable if you are 1) using reads shorter than 100bp, 2) using a tensor with flanking length longer than 16bp, and 3) you are using amplicon sequencing or other sequencing technologies, in which reads starting positions are random is not a basic assumption". |
PairWithNonVariants.py |
Pair truth variant tensors with non-variant tensors. Input: Truth variants tensors; Candidate variant tensors. Important options: --amp x "1-time truth variants + x-time non-variants". |
ChooseItemInBed.py |
Helper script. Output the items overlapping with input the BED file. |
CountNumInBed.py |
Helper script. Count the number of items overlapping with the input BED file. |
param.py |
Global parameters for the scripts in the folder. |
PrepDataBeforeDemo.sh |
A Demo showing how to prepare data for model training. |
PrepDataBeforeDemo.pypy.sh |
The same demo but using pypy in place of python. pypy is highly recommended. It's easy to install, and makes the scripts run 5-10 times faster. |
CombineMultipleDatasetsForTraining.sh |
Use chr21 and chr22 to exemplify how to maunually combine multiple datasets for model training. |
CombineMultipleDatasetsForTraining.py |
A helper script for combining multiple datasets. You still need to run PairWithNonVariants.py and tensor2Bin.py after this script. |
clairvoyante/ |
Model Training and Variant Caller Scripts. Scripts in this folder are NOT compatible with pypy. Please run with python. |
|---|---|
callVar.py |
Call variants using candidate variant tensors. |
callVarBam.py |
Call variants directly from a BAM file. |
callVarBamParallel.py |
Generate callVarBam.py commands that can be run in parallel. A BED file is required to specify the regions for variant calling. --refChunkSize set the genome chuck size per job. |
demoRun.py |
A Demo showing how to train a model from scratch. |
evaluate.py |
Evaluate a model in terms of base change, zygosity, variant type and indel length. |
param.py |
Hyperparameters for model training and other global parameters for the scripts in the folder. |
tensor2Bin.py |
Create a compressed binary tensors file to facilitate and speed up future usage. Input: Mixed tensors by PairWithNonVariants.py; Truth variants by GetTruth.py and a BED file marks the high confidence regions in the reference genome. |
train.py |
Training a model using adaptive learning rate decay. By default, the learning rate will decay for three times. Input a binary tensors file created by Tensor2Bin.py is highly recommended. |
trainNonstop.py |
Helper script. Train a model continuously using the same learning rate and l2 regularization lambda. |
trainWithoutValidationNonstop.py |
Helper script. Train a model continuously using the same learning rate and l2 regularization lambda. Take all the input tensors as training data and do not calculate loss in the validation data. |
calTrainDevDiff.py |
Helper script. Calculate the training loss and validation loss on a trained model. |
getTensorAndLayerPNG.py |
Create high resolution PNG figures to visualize input tensor, layer activations and output. |
getEmbedding.py |
Prepare a folder readable by Tensorboard for visualizing predicted results. |
clairvoyante_v3.py |
Clairvoyante network topology v3. |
clairvoyante_v3_slim.py |
Clairvoyante network topology v3 slim. With ten times fewer parameters than the full network, it trains about 1.5 times faster than the full network. It performs only about 1% less in precision and recall rates for Illumina data. |
utils_v2.py |
Helper functions to the network. |
GIAB provides a BED file that marks the high confidence regions in the reference. The models perform better by using only the truth variants in these regions for training. If you don't have such a BED file, you can use a BED file that covers the whole genome.
The trained models are in the trainedModels/ folder.
| Folder | Tech | Aligner | Ref | Sample |
|---|---|---|---|---|
fullv3-illumina-novoalign-hg001+hg002-hg38 |
Illumina HiSeq25001 | Novoalign 3.02.07 | hg38 | NA12878+NA24385 |
fullv3-illumina-novoalign-hg001-hg38 |
Illumina HiSeq25001 | Novoalign 3.02.07 | hg38 | NA12878 |
fullv3-illumina-novoalign-hg002-hg38 |
Illumina HiSeq25001 | Novoalign 3.02.07 | hg38 | NA24385 |
fullv3-pacbio-ngmlr-hg001+hg002+hg003+hg004-hg19 |
mainly PacBio P6-C42 | NGMLR 0.2.6 | hg19 | NA12878+NA24385+NA24149 +NA24143 |
fullv3-pacbio-ngmlr-hg001+hg002-hg19 |
PacBio P6-C42 | NGMLR 0.2.6 | hg19 | NA12878+NA24385 |
fullv3-pacbio-ngmlr-hg001-hg19 |
PacBio P6-C42 | NGMLR 0.2.6 | hg19 | NA12878 |
fullv3-pacbio-ngmlr-hg002-hg19 |
PacBio P6-C42 | NGMLR 0.2.6 | hg19 | NA24385 |
fullv3-ont-ngmlr-hg001-hg19 |
Oxford Nanopore Minion R9.43 | NGMLR 0.2.6 | hg19 | NA12878 |
1 Also using Illumina TruSeq (LT) DNA PCR-Free Sample Prep Kits. Zook et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. 2016
2 Pendelton et al. Assembly and diploid architecture of an individual human genome via single-molecule technologies. 2015
* Each folder contains one or more models. Each model contains three files suffixed data-00000-of-00001, index and meta, respectively. Only the prefix is needed when using the model with Clairvoyante. Using the prefix learningRate1e-3.epoch999.learningRate1e-4.epoch1499 as an example, it means that the model has trained for 1000 epochs at learning rate 1e-3, then another 500 epochs at learning rate 1e-4. Lambda for L2 regularization was set the same as learning rate.
| Equiptment | Seconds per Epoch per 11M Variant Tensors |
|---|---|
| Tesla V100 | 90 |
| GTX 1080 Ti | 170 |
| GTX 980 | 350 |
| GTX Titan | 520 |
| Tesla K40 (-ac 3004,875) | 580 |
| Tesla K40 | 620 |
| Tesla K80 (one socket) | 600 |
| GTX 680 | 780 |
| Intel Xeon E5-2680 v4 28-core | 2900 |
Different from model training, in which all genome positions are candidates but randomly subsampled for training, variant calling using a trained model will require the user to define a minimal alternative allele frequency cutoff for a genome position to be considered as a candidate for variant calling. For all sequencing technologies, the lower the cutoff, the lower the speed. Setting a cutoff too low will increase the false positive rate significantly, while too high will increase the false negative rate significantly. The option --threshold controls the cutoff in these three scripts callVarBam.py, callVarBamParallel.py and ExtractVariantCandidates.py. The suggested cutoff is listed below for different sequencing technologies. A higher cutoff will increase the accuracy of datasets with poor sequencing quality, while a lower cutoff will increase the sensitivity in applications like clinical research. Setting a lower cutoff and further filter the variants by their quality is also a good practice.
| Sequencing Technology | Alt. AF Cutoff |
|---|---|
| Illumina | 0.125 |
| PacBio P6-C4 | 0.2 |
| ONT R9.4 | 0.25 |
The testing dataset 'testingData.tar' includes:
- the Illumina alignments of chr21 and chr22 on GRCh38 from GIAB Github, downsampled to 50x.
- the truth variants v3.3.2 from GIAB.
Clairvoyante network version 3 can only output one of the two possible alternative alleles at a position. We will further extend the network to support genome variants with two alternative alleles.
In rare cases, the model training will stuck early at a local optimal and cannot be further optimized without a higher learning rate. As we observed ,the problem only happens at the very beginning of model training, and can be predicated if the loss remains stable in the first few training epochs.