zzAttic Design document
Scalable datastore for population genome sequencing, with on-demand joint genotyping (GL, genotype likelihood)
This is an early-stage DNAnexus R&D project we're developing openly. The code doesn't yet do anything useful! The following roadmap should be read in the spirit of "plans are worthless, but planning is indispensable."
GLnexus is a new component in data processing pipelines for high-throughput genome sequencing, supporting collection of small genetic variants across millions of whole human genomes. It's a scalable solution for certain data aggregation steps which sit downstream of raw data crunching on short reads, but upstream of statistical analysis and querying on completed genotype datasets. These steps, which can be oversimply described as "VCF merging", include:
- unifying the representation of related sequence variants sampled in the population
- empirically updating Bayesian priors informing joint genotyping
- generating a population-wide joint genotype matrix in a form convenient for statistical analysis
Operationally, GLnexus is a datastore intended for use in a large-scale project engaged in sequencing many samples over months or years. You deposit gVCF data for individual samples continuously as they're sequenced, and from time to time extract multi-sample VCF data derived from the samples deposited up to that point. The name GLnexus derives from genotype likelihoods being the key intermediate data conveyed in each gVCF. For current users of the GATK, GLnexus can be viewed roughly as a scalable replacement for CombineGVCFs and GenotypeGVCFs, although its design includes numerous additional functions. (We gladly cite allusions to a "database of likelihoods" in GATK docs, probably a similar concept.)

The gVCF and VCF data models are pragmatically useful for now, but may be replaced or augmented in the future, e.g. with unitig alignments or graph abstractions. More important is that we reframe joint genotyping as a malleable way of "rendering" a multifaceted corpus of variation data, instead of an epic batch job producing the canonical results. By analogy to a 2D rendering of a 3D scene, this acknowledges that not all of the intermediate data can be faithfully captured in the multi-sample VCF data model, and different downstream applications and analyses may call for the data corpus to be "rendered" from different angles.
GLnexus has been motivated by DNAnexus' experience serving our large-scale customers/partners as they contemplate future million-plus genome projects. Given cloud-scale computing resources, the raw data crunching which would occur upstream of GLnexus, embarassingly parallel across samples, looks quite tractable with existing methods (e.g. read mapping and variant calling up to gVCF). Meanwhile, scalable approaches for statistical analysis/query of genotype datasets, downstream of GLnexus, are being pursued in diverse projects such as PLINK 2, gqt, bgt, and various applications of generic scale-out data warehouses. The challenges we aim to address here appear underserved in contrast.
To generate multi-sample VCF/BCF data for a specified region of the reference genome, GLnexus executes the following steps. This is probably very similar to GenotypeGVCFs, although we're steadfastly avoiding its source code.
- Discover alleles. Collect all distinct alleles overlapping the reference region, each represented by reference range and DNA sequence. Also accumulate an observation count for each allele, and heuristically retain common read-based phase sets.
- Delineate sites. Partition the alleles into non-overlapping "sites". Prune alleles causing excessive collapsing of otherwise-distinct sites based on some heuristics (e.g. rarity vs. length)
- Unify alleles. Within each delineated site, pad the individual alleles with reference bases so that they all cover the same reference range. Join nearby alleles, when possible, with read-based phase sets. Use the hard observation counts to make empirical estimates of the population allele frequencies. Prune rare alleles, if necessary to keep the number of possible genotypes under control, as well as alleles that are problematic to unify under our constraints.
- Genotype samples at each unified site using a Bayesian calculation on the estimated allele frequencies (⇒ genotype priors), original GLs of the alleles in each gVCF, and a gVCF-based reference likelihood model for the additional bases in padded alleles. Render any allele that was pruned in the global unification as some appropriate symbolic allele.
- Render VCF, by which we mean serialize the results into the logical VCF/BCF data model and export to some other environment. (It may or may not make sense to create a "VCF file" per se.)
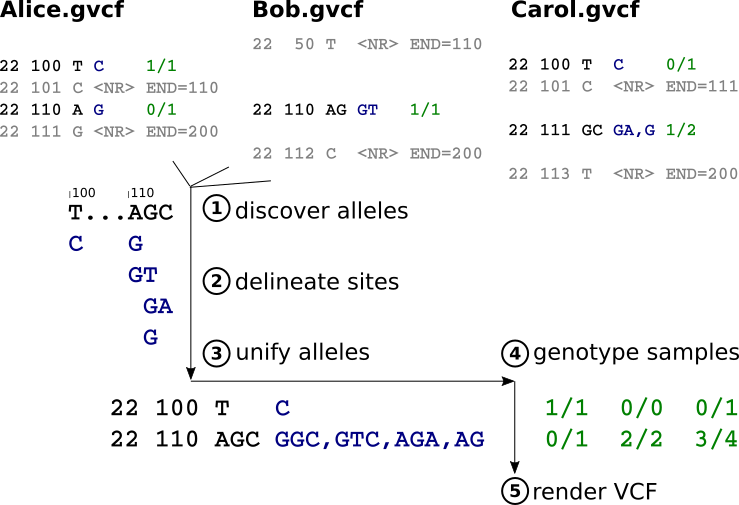
Step 1 is a map-reduce over samples (gVCFs). Steps 2 & 3 are serial but fast as they deal only with sample-independent allele lists. Step 4 is embarassingly parallel over samples and/or sites. The parallelism of step 5 depends on what the 'other environment' is. The whole process is embarassingly parallel over disjoint genomic regions.
JOINT LIKELIHOOD FORMULAE GOES HERE
FURTHER EXAMPLES GO HERE
Observations/ideas:
- We can cache the unified sites for a set of samples. And most of the time, we can incrementally compute the sites for a superset from the cached sites and the alleles in the additional samples.
- We may derive unified alleles from a different set of samples than we actually genotype (i.e. a reference panel), or the request may even supply a desired allele set.
- The request may designate a set of 'focal' samples whose original alleles we're reluctant to prune during unification, even if they're very rare.
- The request may include a pedigree for a subset of samples to structure the genotype prior. For example, the priors for a focal trio can be structured to simultaneously incorporate population allele frequencies and to penalize Mendelian inconsistencies.
- Further elaboration of the allele unification algorithm seems likely to converge to a local graph model, in the fullness of time - but we'll start with the simpler linear approach outlined above, and prune what we can't handle.
- Speculative future directions: phase and impute, infer consequences, generally solve all other problems in genetics
We'd like to store our gVCF corpus in a way that enables us to efficiently (a) slice out a given genomic range across samples and (b) add new sample gVCFs continuously over time. Here's where we'll initially make some concessions to simplicity and wheel-nonreinvention. We store gVCF data using the VCF/BCF data model as implemented in htslib - but not in a pile of gVCF files. Instead, we MacGyver htslib to divorce the serialization of the data model (bcf1_t) from the block-compressed container format (BGZF with header).
We can then serialize the gVCF records into a different storage medium better supporting the aforementioned slicing operation. We're initially trying out a write-optimized, ordered key-value store. We quite like RocksDB, which is a local storage library, but "cloud databases" like DynamoDB or Cassandra are also plausible.
Each input sample gVCF file is split up into pieces based on predetermined genomic range buckets. Each piece, containing one or more individual gVCF records, is keyed by the tuple of (bucket,sample) and inserted into the ordered key-value database. Records spanning multiple genomic range buckets are duplicated in each bucket. All the gVCF headers are squirreled away in some other part of the key space.

To slice a genomic range across samples, we look up the overlapping buckets and iterate over the respective contiguous ranges of the key space, parsing the gVCF header and records into memory. Efficient ingestion of new gVCFs into the ordered key space is up to the database, which had better use LSM or a similar technique.
Observations/ideas:
- The gVCF ingestion process should include indel left-alignment and other simple normalizations.
- Consolidation of multiple samples into individual values (cf. CombineGVCFs) could be advantageous under some circumstances.
- We could furthermore make selection of samples by ethnicity (or some other attribute) more efficient by formulating keys like (bucket,ethnicity,sample).
To scale out across compute nodes, genotyping operations can be decomposed and parallelized along two dimensions: shards of the sample set on the one hand, and non-overlapping genomic regions on the other hand. If the gVCF data are appropriately sharded on the compute nodes, or stored in a "cloud database" abstracting us from the storage locality, then the genotyping algorithm steps outlined above require very little movement of intermediate data between nodes - essentially just allele lists. (The final genotype matrix must be retrieved of course, and this is why it may not always be preferable to produce a "VCF file" in the end.)
We haven't yet decided on the framework for orchestrating the distributed genotyping operations. Given the modest data movement needs, a lot of different frameworks could work here. Our first thought is to have a coordinator node that makes REST/Thrift/gRPC remote procedure calls to other nodes to discover alleles and then request genotyping of unified sites; the delineation and unification just happens on the coordinator node. We'll bake as much core functionality as possible into this C++ library (libglnexus), so the orchestration mechanism could even be modularized.
The web-friendly RPC protocol we have postulated immediately suggests a possible non-copying mechanism for federated variant discovery and genotyping across projects/institutions/clouds.
- Is joint genotyping truly necessary in the era of cheap high-coverage WES/WGS? Note, the concept encompasses at least three separable pillars:
- Allele unification and "squaring off" the genotype matrix
- Updating estimates of population allele frequencies (priors)
- Modeling and compensating for systematic errors
- Is someone else already building something like this, with which we'd be able to serve our customers/partners?
- Will adopting the [g]VCF data model grant us a solid window of valuable service, before fundamentally different paradigms (e.g. reference graph, [long] read-based phasing/SV) are ready for population-scale projects?
- Is our factorization of the genotyping algorithm missing cross-site dependencies in the likelihood model, or any other key dimensions of the problem?