

Predict (tissue-specific) splicing variant effect from VCF. MTSplice is integrated into MMSplice with the same API.
Paper: Cheng et al. https://doi.org/10.1101/438986, https://www.biorxiv.org/content/10.1101/2020.06.07.138453v1
External dependencies:
pip install cyvcf2 cythonConda installation is recommended:
conda install cyvcf2 cython -ypip install mmspliceYou can run mmsplice with following google colab notebooks online:
Standard human gene annotation file in GTF format can be downloaded from ensembl or gencode.
MMSplice can work directly with those files, however, some filtering is higly recommended.
- Filter for protein coding genes.
A correctly formatted VCF file with work with MMSplice, however the following steps will make it less prone to false positives:
- Quality filtering. Low quality variants leads to unreliable predictions.
- Avoid presenting multiple variants in one line by splitting them into multiple lines. Example code to do it:
bcftools norm -m-both -o out.vcf in.vcf.gz
- Left-normalization. For instance, GGCA-->GG is not left-normalized while GCA-->G is. Details for unified representation of genetic variants see Tan et al.
bcftools norm -f reference.fasta -o out.vcf in.vcf
Human reference fasta file can be downloaded from ensembl/gencode. Make sure the chromosome name matches with GTF annotation file you use.
Check notebooks/example.ipynb
To score variants (including indels), we suggest to use primarily the deltaLogitPSI predictions, which is the default output. The differential splicing efficiency (dse) model was trained from MMSplice modules and exonic variants from MaPSy, thus only the predictions for exonic variants are calibrated.
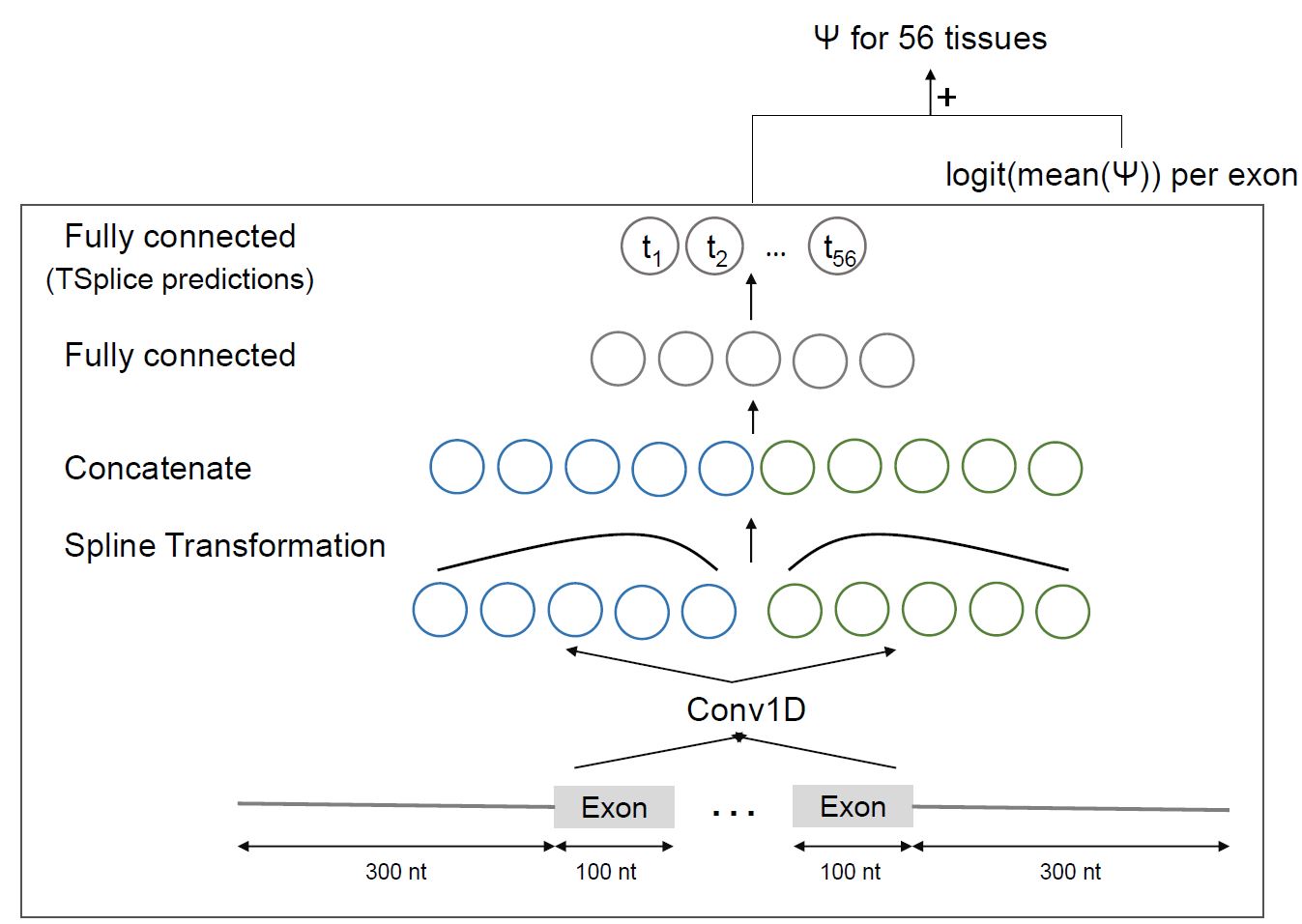
MTSplice To predict tissue-specific variant effect with MTSplice, specify tissue_specific=True in SplicingVCFDataloader.
# Import
from mmsplice.vcf_dataloader import SplicingVCFDataloader
from mmsplice import MMSplice, predict_save, predict_all_table
from mmsplice.utils import max_varEff
# example files
gtf = 'tests/data/test.gtf'
vcf = 'tests/data/test.vcf.gz'
fasta = 'tests/data/hg19.nochr.chr17.fa'
csv = 'pred.csv'Dataloader to load variants from vcf
dl = SplicingVCFDataloader(gtf, fasta, vcf, tissue_specific=False)To predict tissue-specific effect, in the dataloader use tissue_specific=True in the dataloader instead
dl = SplicingVCFDataloader(gtf, fasta, vcf, tissue_specific=True)Run prediction with default MMSplice parameters
# Specify model
model = MMSplice()
# Or predict and return as df
predictions = predict_all_table(model, dl, pathogenicity=True, splicing_efficiency=True)To predict variant effect on scale instead of
. This option only works with tissue specific predictions
dl = SplicingVCFDataloader(..., tissue_specific=True):
# Or predict and return as df
predictions = predict_all_table(model, dl, natural_scale=True)One variant might map to multiple exons. In the end we summarize the effect of as the maximum across all exons.
# Summerize with maximum effect size
predictionsMax = max_varEff(predictions)Output of MMSplice is an tabular data which contains following described columns:
ID: id string of the variantdelta_logit_psi: The main score is predicted by MMSplice, which shows the effect of the variant on the inclusion level (PSI percent spliced in) of the exon. The score is on a logit scale. If the score is positive, it shows that variant leads higher inclusion rate for the exon. If the score is negative, it shows that variant leads higher exclusion rate for the exon. If delta_logit_psi is bigger than 2 or smaller than -2, the effect of variant can be considered strong.exons: Genetics location of exon whose inclusion rate is effected by variantexon_id: Genetic id of exon whose inclusion rate is effected by variantgene_id: Genetic id of the gene which the exon belongs to.gene_name: Name of the gene which the exon belongs to.transcript_id: Genetic id of the transcript which the exon belongs to.ref_acceptorIntron: acceptor intron score of the reference sequenceref_acceptor: acceptor score of the reference sequenceref_exon: exon score of the reference sequenceref_donor: donor score of the reference sequenceref_donorIntron: donor intron score of the reference sequencealt_acceptorIntron: acceptor intron score of variant sequencealt_acceptor: acceptor score of the sequence with variantalt_exon: exon score of the sequence with variantalt_donor: donor score of the sequence with variantalt_donorIntron: donor intron score of the sequence with variantpathogenicity: Potential pathogenic effect of the variant.efficiency: The effect of the variant on the splicing efficiency of the exon.
The VEP plugin wraps the prediction function from mmsplice python package. Please check documentation of vep plugin under VEP_plugin/README.md.