Paired end reads linkage in BAM and CRAM file #521
Comments
|
Do you have a link for how this looks? I don't think we have this feature http://www.broadinstitute.org/igv/view_as_pairs You could however, color by template length, which might give you some ideas for how the paired end reads are linked in a region. You can do this by using the style.color as a javascript callback (short mailing list thread here with example http://sourceforge.net/p/gmod/mailman/message/32619454/) Other links |
|
Note that "template_length" would be used in place of "length_on_ref" RE: http://sourceforge.net/p/gmod/mailman/message/32619454/ |
|
Hi Colin, We should discuss this because we -did- have a means of doing this that And the data needs to use a standardized method for indicating the pairs so -S On Fri, Oct 17, 2014 at 8:22 AM, Colin Diesh notifications@github.com
|
|
So is that means currently jbrowse still does not have solution on visualizing paired end strands connected each other similar with IGV? |
|
@Kinztan I don't think so although I haven't used IGV with paired end reads. There are some other features in jbrowse that can sort of help with this however. For example, paired end reads are colored based on whether they are properly paired or whether there is aberrant pairing in jbrowse. This helps you quickly identify the reads that have strange pairing patterns (no mate pair, or abberant mate pair) and can call your attention. We may also have a further discussion about this as @selewis said |
|
Some discussion that was sort of tangential was moved to here #560 |
|
@scottcain you have some paired reads data that you have been working with at wormbase, could you maybe paste your configuration here? |
|
@rbuels, as much as I'd like for it to be generally applicable to BAM read pairs, the work I've done with pair ESTs is sufficiently different as to not be very helpful. What I've done hinges on preprocessing the EST match GFF and creating a parent "experimental_feature" feature that has the two match features as children. So it looks kind of like this: The only way I could see this working for BAM data would be if there were a process in JBrowse that would do the same sort of thing, but I have a hard time believing it would be anywhere near fast enough. If it's helpful at all, the glyph I wrote is here: |
|
This is a code for connecting bam reads that adds two new glyphs, one using arcs and another with lines https://github.com/elsiklab/apollo_lsaa/tree/master/client/PairedReadViewer It uses the bam coordinate position of the mate pairs to just draw lines "towards" it's mate |
|
@rbuels , WGS paired end data available here: ftp://ngs.sanger.ac.uk/production/cancer/dockstore/cgpwgs/ Corresponding ref is Human GRCh37 (no chr prefix) |
|
Oh thanks so much for that! Good sample data is key.
…On Fri, Feb 2, 2018 at 3:16 AM Keiran Raine ***@***.***> wrote:
@rbuels <https://github.com/rbuels> , WGS paired end data available here:
ftp://ngs.sanger.ac.uk/production/cancer/dockstore/cgpwgs/
Corresponding ref is Human GRCh37 (no chr prefix)
—
You are receiving this because you were mentioned.
Reply to this email directly, view it on GitHub
<#521 (comment)>, or mute
the thread
<https://github.com/notifications/unsubscribe-auth/AAEgFWvDHw8u0sNoFk2Ic5LaKsFLA6s2ks5tQu6YgaJpZM4Cv2AK>
.
|
|
implementation sketch
|
|
The IGV concept of "view as pairs" is probably sort of the idea we want to get at. They have this feature in igv js code here https://github.com/igvteam/igv.js/blob/master/js/bam/bamSource.js An igv screenshot shows the idea, especially related to revealing structural issues like deletion it is great. screenshot is near bottom of page here https://bioinformaticsdotca.github.io/BiCG_2017_module2_igv |
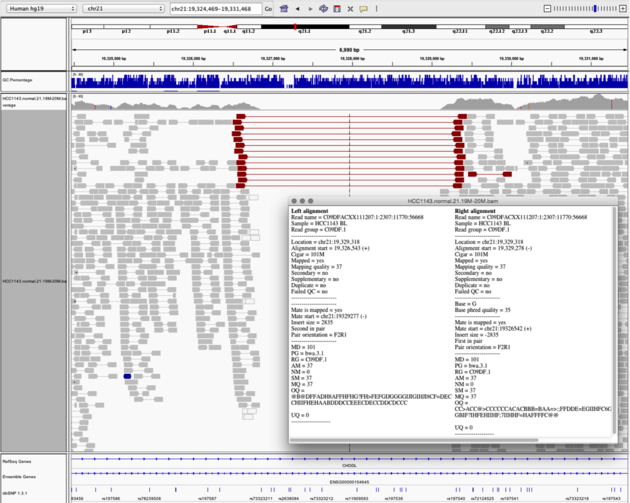
|
A cursory look at the igvjs code looks like it is a bit different from the block based rendering that jbrowse uses. It gets a large "alignmentContainer" that gets all alignments within the current view plus a bit outside the boundaries of the view too. Then, when you scroll sideways far enough, it will then reload the "alignmentContainer" with new reads, and redo pairings. This type of strategy is something that can probably be emulated with the JBrowse codebase but it would be sort of a different strategy than the block based style track/store that is used in most tracks. |
|
What if we do something similar to GFFTabix, where it redispatches requests
if paired is on? Would be simplest to implement, and might not actually be
as slow as one might think, if the feature caching is working well.
…On Sat, Jul 21, 2018 at 9:30 AM Colin Diesh ***@***.***> wrote:
A cursory look at the igvjs code looks like it is a bit different from the
block based rendering that jbrowse uses. It gets a large
"alignmentContainer" that gets all alignments within the current view plus
a bit outside the boundaries of the view too. Then, when you scroll
sideways far enough, it will then reload the "alignmentContainer" with new
reads, and redo pairings. This type of strategy is something that can
probably be emulated with the JBrowse codebase but it would be sort of a
different strategy than the block based style track/store that is used in
most tracks.
—
You are receiving this because you were mentioned.
Reply to this email directly, view it on GitHub
<#521 (comment)>, or mute
the thread
<https://github.com/notifications/unsubscribe-auth/AAEgFWCwzeiep3xgwJ3YUW9B5aPZld-Jks5uI1bugaJpZM4Cv2AK>
.
|
|
Nice idea. I'll see what that might entail...it sounds pretty reasonable |
|
At least one scenario that is difficult when just looking at a small block-by-block basis is if a BAM pair is on either side of a block, then the block will basically have no knowledge of it. Example, this view works at a zoomed out level with several pairs going across the deletion, but if you zoom in, such that you are inside the deletion, it looks glitchy. Here's the nice non glitchy screenshot though :) |

|
The alignmentContainer thing would be basically making the track one big glyph--that way everything knows about everything else in the view (and a little nearby, I imagine). It wouldn't be too hard to implement; it would be similar to what I did to make the paired ESTs work (though I built the "super object" by preprocessing the GFF). It would certainly work, but would it be performant? |
|
Another thing that would be nice about the track being a single large glyph is that you'd be able to do things that are typically hard to do in a track, like sorting on arbitrary things: are they paired or not, are there splice junctions or not. Could make for a really nice display, putting the "important" things at the top of the track. |
|
We reviewed example read pairing code from PairedReadViewer and looked at what our steps will probably be for obtaining solid functionality for this. Steps are probably as follows step 1 refactor remotebinarystore with query aggregation |
|
This was delivered in JBrowse 1.16.0! Please give it a try and make new bugs if there are any issues or suggestions |
|
See docs here http://jbrowse.org/docs/paired_reads.html and blogpost http://jbrowse.org/blog/2018/12/13/jbrowse-1-16-0.html |
|
Hi there! Is this feature available on JBrowse2? It would be a very useful addition. |
|
@malonge we recently added "Arc view" and "Read cloud" view to jbrowse 2, which helps show paired reads as linked entities Some info here https://jbrowse.org/jb2/blog/2022/12/15/v2.3.1-release/ It doesn't draw the pairs using the "stacked layout" like you'd normally see, but instead draws arcs (for the "Arc view") or lays them out stratified by "insert size" on the y-axis (for the "Read cloud") We may add the normal "stacked" layout as a feature in the future, but hopefully this helps :) Some example links
|
|
awesome thank you! looks like I need to update to a newer version of JBrowse2 (I am still using v2.0.1). Thanks! |
Is there any function that can enable to visualize the paired end links between two pair ends. Just like the broad IGV did, it is quite important to visualize the linkage of pair end sequence to understand their mechanism. Is there any configuration I need to change for that?
The text was updated successfully, but these errors were encountered: