Tutorial: https://interactivereport.github.io/scRNAsequest/tutorial/docs/index.html
Publication: https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-023-09332-2
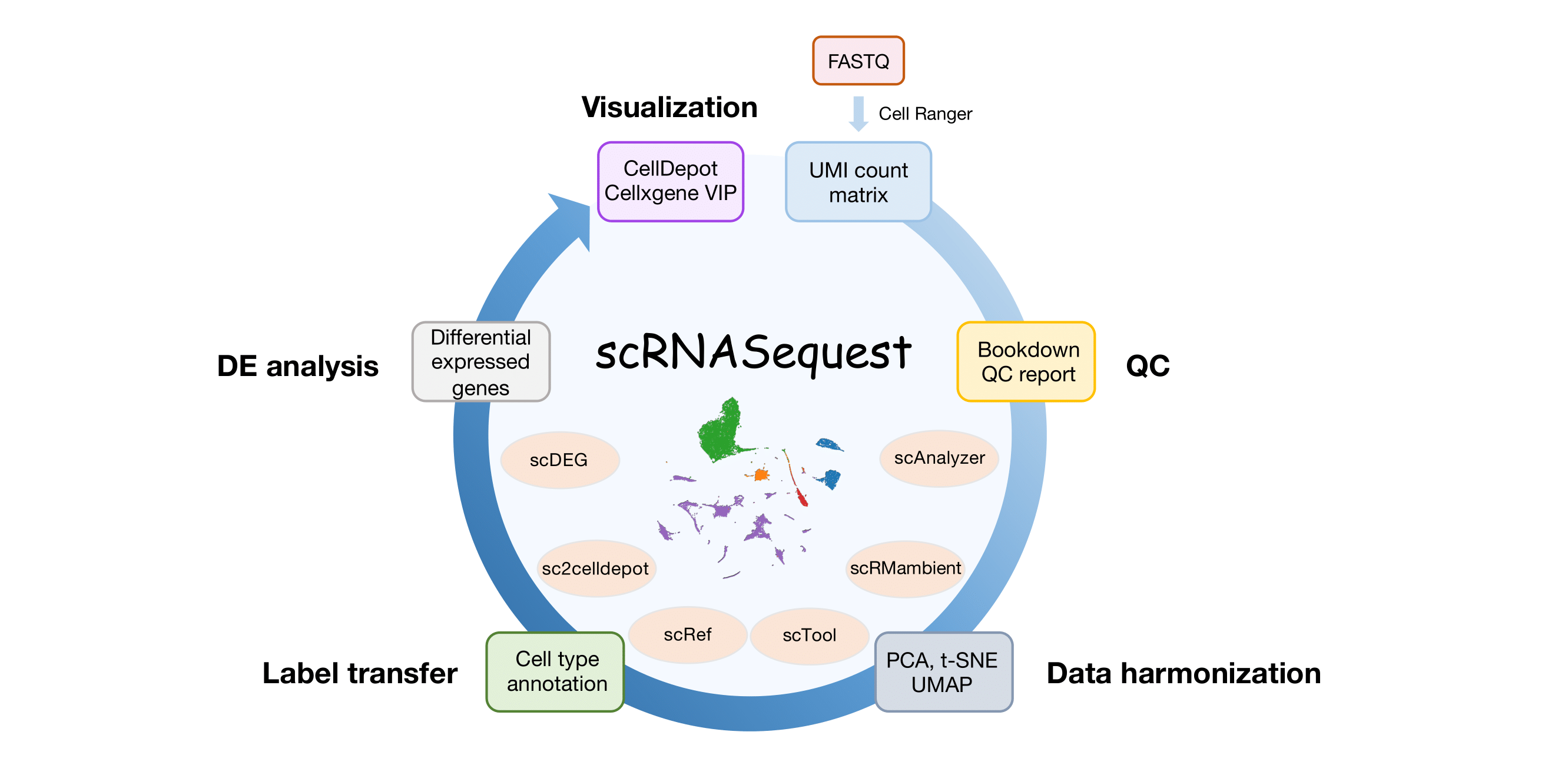
Fig. 1. Overview of the scRNASequest workflow. User provides single-cell or single-nucleus RNA-seq gene expression matrix (h5 or MEX) from Cell Ranger and sample metadata to the semi-automated workflow, scRNASequest. It generates basic quality control (QC) Bookdown reports and allows users to choose from popular data harmonization tools such as Harmony, LIGER, and Seurat to remove batch effects. Azimuth reference-based cell label transfer is enabled as optional to perform cell type identification. Cluster- or cell-type-specific multi-sample multi-condition single cell level DE analysis is by default performed through NEBULA. Finally, an h5ad file will be generated to be loaded into the cellxgene VIP framework or CellDepot single-cell data management system for interactive visualization and analysis. The main script for the analysis is scAnalyzer. Five additional scripts are also included in the scRNASequest pipeline suite: scRMambient, scTool, scRef, sc2celldepot, and scDEG.
We provide two methods to install scRNASequest. The first one uses Conda, and the second one uses Docker. We have tested both methods on Linux servers; however, if you are a Mac user, please use the Docker method.
First, please make sure you have Conda installed, or, Anaconda/Miniconda installed:
which conda
# Your conda path will be returned
Then, we choose a directory and install scRNASequest by downloading the source code from GitHub.
The directory you choose here will be the future directory of this pipeline.
# Go to the directory you choose. This tutorial uses $HOME (~) directory as an example:
cd ~
git clone https://github.com/interactivereport/scRNASequest.git
cd scRNASequest
# Install scRNASequest conda environment
# Before running this, please make sure you have conda installed before
# This step will take a while, usually between 30min to 1h depending on the internet speed
# Thank you for your patience
bash install.sh
# The .env will be created under the src directory
ls ~/scRNASequest/src/.env
# Now the pipeline scripts under the scRNASequest folder can be used
# Users can add the scRNASequest directory to the environment permanently
# by editing ~/.bash_profile or ~/.bashrc
vim ~/.bash_profile
# Add the full path of the scRNASequest directory to $PATH.
# In our example, this will be: ~/scRNASequest
PATH=$PATH:~/scRNASequest
# Close the vim text editor and source the file
source ~/.bash_profile
#To verify the installation, type the main program name, and the following message will show up:
scAnalyzer
#Output:
=====
Please set the sys.yml in ~/scRNASequest.
An example is '~/scRNASequest/src/sys_example.yml'.
=====
You got the above message because the sys.yml file is missing under the pipeline src directory (in our case, ~/scRNASequest/src/).
Please copy the sys_example.yml template there first:
cp ~/scRNASequest/src/sys_example.yml ~/scRNASequest/src/sys.yml
Then, fill in the following required items. These directories will be used to host final results (.h5ad files) of the pipeline as well as the reference files for cell type label transfer.
Since we have cellxgenedir and ref directories created under the demo directory, we use them here:
celldepotDir: ~/scRNASequest/demo/cellxgenedir. # the absolute path to the cellxgene VIP host folder, where the h5ad files will be copied to for cellxgene VIP
refDir: ~/scRNASequest/demo/ref # the absolute path to the Seurat reference folder if building reference is desired
You may fill in the Cellxgene VIP server path after installing Cellxgene VIP later, but this is not required for running the pipeline. We leave this empty here.
celldepotHttp: # the cellxgene host (with --dataroot option) link http://HOST:PORT/d/
You may change the information in the sys.yml file later, following the full tutorial here.
Then type the name of the main program, scAnalyzer again:
***** 2023-03-14 14:48:13 *****
###########
## scRNAsequest: https://github.com/interactivereport/scRNAsequest.git
## Pipeline Path: /mnt/depts/dept04/compbio/projects/ndru_projects/Software/scRNAsequest
## Pipeline Date: 2023-03-01 10:10:55 -0500
## git HEAD: d067bfd6dc056597d046a45f3b3b927dd122dd82
###########
scAnalyzer /path/to/a/DNAnexus/download/folder === or === scAnalyzer /path/to/a/config/file
The config file will be generated automatically when a DNAnexus download folder is provided
Available reference data:
human_cortex: more information @ https://azimuth.hubmapconsortium.org/references/
If one of the above can be used as a reference for your datasets, please update the config file with the name in 'ref_name'.
Powered by None
------------
The installation was successful if you see the above message.
We provide a Docker image here: https://hub.docker.com/repository/docker/sunyumail93/scrnasequest/general. Users can pull this image to build a container, which has been tested on both Linux and Mac systems. This will take roughly 10 minutes to set up.
We also provide a Dockerfile if you would like to build the image from scratch using the docker build command, which takes ~30 min.
First, please make sure Docker has been installed and can be recognized through the command line:
which docker
# Your docker path will be returned
Go to the directory you choose. This tutorial uses $HOME (~) directory as an example:
cd ~
git clone https://github.com/interactivereport/scRNASequest.git
cd scRNASequest
Then we pull the docker image. This step takes ~10 min.
docker pull sunyumail93/scrnasequest
Initiate the docker container. This command uses -v to map the demo directory under scRNASequest to /demo in the container:
docker run -v `pwd`/demo:/demo -d sunyumail93/scrnasequest
The above command prepars for the demo run in section 2.2. You can mount any directory containing your data to the Docker container using the syntax old_dir:container_dir.
Verify your container:
docker container ls
#Results:
CONTAINER ID IMAGE COMMAND CREATED STATUS PORTS NAMES
4e0f3a40ce1d sunyumail93/scrnasequest "/bin/sh -c 'while t…" 54 seconds ago Up 52 seconds interesting_lewin
The last column is the <container_name>, and it will be used in the following steps.
Now we launch the main program of this pipeline. In our example, <container_name> is interesting_lewin. Please substitute to yours:
docker exec -t -i <container_name> scAnalyzer
#Output:
=====
Please set the sys.yml in /home/scRNASequest/src.
An example is '/home/scRNASequest/src/sys_example.yml'.
=====
This is because the sys.yml configuration file is missing under the src directory. There is a sys.yml file prepared for running the demo data (see section 2.2), and you can copy it to the pipeline src directory using the command below. It will work for future analysis also. However, you may change the information in the sys.yml file later, following the full tutorial here. Please note that both '/demo/sys.yml' and '/home/scRNASequest/src' directories are in the container, rather than in your file system.
docker exec -t -i <container_name> cp /demo/sys.yml /home/scRNASequest/src/
Now we run this command again, and we will see the pipeline message printed out, same as the one at the end of section 1.1:
docker exec -t -i <container_name> scAnalyzer
We provide a demo dataset under the demo directory. This demo uses two snRNA-seq data from GSE185538 to run through the main steps, including QC, data integration (SCTransform, then Harmony), Seurat reference mapping, and evaluation of integration (kBET and silhouette). To save time, the differential expression analysis won't be performed, and the DEGinfo.csv file is empty.
This demo run contains two downsampled snRNA-seq data from the original study, and will take ~15-20 minutes to finish. Please note that to speed up the run, we used stringent QC cutoffs, which eliminated many cells. After running the scAnalyzer command, output files will be generated under the demo directory on your computer.
Also, we provided config.yml, sampleMeta.csv and DEGinfo.csv files for running this demo so you can run the scAnalyzer command directly. For your own dataset, please follow section 3.2 to generate the template of these files first.
All necessary files for this demo have been prepared under the demo directory.
Continuing from section 1.1, we assume that the pipeline was installed in: ~/scRNASequest.
First, we set up the config file (~/scRNASequest/demo/config.yml) by pointing these parameters to your directory because the default directory was /demo, and other lines don't need to be changed:
...
ref_name: ~/scRNASequest/demo/ref/rat_cortex_ref.rds # choose one from scAnalyzer call without argument
output: ~/scRNASequest/demo # output path
...
sample_meta: ~/scRNASequest/demo/sampleMeta.csv
...
DEG_desp: ~/scRNASequest/demo/DEGinfo.csv # for DEG analysis
...
Accordingly, we modify the sampleMeta.csv file by providing the full path to .h5 files by adding '~/scRNASequest':
Sample_Name,h5path,Sex
RatFemaleCigarette,~/scRNASequest/demo/data/RatFemaleCigarette.filtered_feature_bc_matrix.h5,Female
RatMaleCigarette,~/scRNASequest/demo/data/RatMaleCigarette.filtered_feature_bc_matrix.h5,Male
Then execute the following command:
scAnalyzer ~/scRNASequest/demo/config.yml
After following section 1.2 to set up the pipeline, we are ready to run this demo:
docker exec -t -i <container_name> scAnalyzer /demo/config.yml
This is a quick start guide to run your own data using the pipeline. Please refer to the full tutorial for more details.
This pipeline accepts h5 or MTX (an mtx file and associated barcodes file as well as a features file) containing cell count information after running Cell Ranger. You can also include the Cell Ranger QC results: DataPrefix.metrics_summary.csv, but this is optional. When you use downloaded data from NCBI/GEO, it may be necessary to rename the files.
An example of h5 input file hierarchy, using data from E-MTAB-11115:
E-MTAB-11115/
├── 5705STDY8058280.raw_feature_bc_matrix.h5
├── 5705STDY8058280.metrics_summary.csv
├── 5705STDY8058281.raw_feature_bc_matrix.h5
├── 5705STDY8058281.metrics_summary.csv
├── 5705STDY8058282.raw_feature_bc_matrix.h5
├── 5705STDY8058282.metrics_summary.csv
├── 5705STDY8058283.raw_feature_bc_matrix.h5
├── 5705STDY8058283.metrics_summary.csv
├── 5705STDY8058284.raw_feature_bc_matrix.h5
├── 5705STDY8058284.metrics_summary.csv
├── 5705STDY8058285.raw_feature_bc_matrix.h5
└── 5705STDY8058285.metrics_summary.csv
An example of MTX input file hierarchy, using data from GSE185538. Files under each separate directory must follow the naming criteria: barcodes.tsv.gz, features.tsv.gz, and matrix.mtx.gz:
GSE185538/
├── GSM5617891_snRNA_FCtr
├── barcodes.tsv.gz
├── features.tsv.gz
└── matrix.mtx.gz
├── GSM5617892_snRNA_FEcig
├── barcodes.tsv.gz
├── features.tsv.gz
└── matrix.mtx.gz
├── GSM5617893_snRNA_MCtr
├── barcodes.tsv.gz
├── features.tsv.gz
└── matrix.mtx.gz
└── GSM5617894_snRNA_MEcig
├── barcodes.tsv.gz
├── features.tsv.gz
└── matrix.mtx.gz
Users can initiate the pipeline by running the scAnalyzer script with a working directory, where future outputs will be generated.
#Example:
scAnalyzer ~/Working_dir
The output files include: config.yml (a template config file), DEGinfo.csv (a template for differential expression analysis), and an empty sampleMeta.csv file. The config.yml and sampleMeta.csv are required to run the pipeline, and the DEGinfo.csv is only required for the DE analysis. You can leave DEGinfo.csv empty (by default, just a header line there) currently.
The empty sampleMeta.csv file contains three preset column headers that can be recognized by the pipeline:
- Sample_Name is used to provide a simplified sample name.
- h5path is for the full data path. If the inputs are h5 files, this would be the full path of each file. However, for MTX inputs, this should be the path to the data directory, where three data files are stored.
- metapath is for cell annotation information, which is optional (See more details about the annotation file). If you don't have this information, you can delete it from the sampleMeta.csv
Users can add more meta information to this sampleMeta.csv. Here are two examples:
#Example 1, for h5 data
Sample_Name,h5path,Sex,Age
5705STDY8058280,~/E-MTAB-11115/5705STDY8058280_filtered_feature_bc_matrix.h5,Female,56d
5705STDY8058281,~/E-MTAB-11115/5705STDY8058281_filtered_feature_bc_matrix.h5,Female,56d
5705STDY8058282,~/E-MTAB-11115/5705STDY8058282_filtered_feature_bc_matrix.h5,Female,56d
5705STDY8058283,~/E-MTAB-11115/5705STDY8058283_filtered_feature_bc_matrix.h5,Male,56d
5705STDY8058284,~/E-MTAB-11115/5705STDY8058284_filtered_feature_bc_matrix.h5,Male,56d
5705STDY8058285,~/E-MTAB-11115/5705STDY8058285_filtered_feature_bc_matrix.h5,Male,56d
#Example 2, for MTX data
Sample_Name,h5path,Treatment,Sex
FCtr,~/GSE185538/GSM5617891_snRNA_FCtr,Control,Female
FEcig,~/GSE185538/GSM5617892_snRNA_FEcig,EcTreated,Female
MCtr,~/GSE185538/GSM5617893_snRNA_MCtr,Control,Male
MEcig,~/GSE185538/GSM5617894_snRNA_MEcig,EcTreated,Male
This config.yml file contains critical configuration parameters to run the pipeline. Please use the following template as an example to prepare this file: config.yml.
Some tips:
- ref_name: If left blank, the pipeline won't run reference-based cell type annotation. Users can provide an RDS or H5ad file following Azimuth reference file format or download from Azimuth references website. To build a reference, use the
scRefscript from our pipeline (See more details). - gene_group: You can define your own gene groups to run QC. If the "rm: False" is set to False, gene counts will be checked, and cells will be filtered out based on the "cutoff" percentage. To completely eliminate contaminating genes, such as mitochondria genes, set "rm: True".
- runAnalysis: False: Run analysis means performing data integration and DE analysis. If set to False, the pipeline will only run the initial QC step, which allows users to examine whether the default cell filtering cutoffs are adequate. If they look good, set it to True to run the whole pipeline.
- overwrite: False: Set to True if you rerun the pipeline and want to overwrite the previous results.
- DEG_desp: Path to the DE configuration file. If the file is empty, it won't perform any analysis. See section 3 about how to fill in this file.
Now we have prepared the minimal files (Data files, sampleMeta.csv, and config.yml) to start the pipeline.
- a. Initiate the analysis with
runAnalysisin config.yml set to False
Here, we pass the path to the config.yml file to run the pipeline:
scAnalyzer ~/Working_dir/config.yml
This will only run the QC step and generate a Bookdown report here.
- b. Examine the QC parameters
Check the Bookdown report and adjust the filtering parameters if needed. Repeat this step until the filtering criteria are satisfied.
- c. Run the full analysis
#Set config.yml:
#runAnalysis: True
#overwrite: True
#Run the full analysis
scAnalyzer ~/Working_dir/config.yml
After running the above steps, you will see a series of files generated. The main results include:
outputdir
├── QC/ # QC plots
├── raw/ # Raw h5ad files before and after cell filtering
├── Liger/ # Liger results
├── sctHarmony/ # Harmony results
├── SeuratRef/ # Seurat results
├── SeuratRPCA/ # SeuratRPCA results
├── evaluation/ # kBET and Silhouette plots
├── project_name_BookdownReport.tar.gz # Bookdown report
├── project_name.h5ad # Final h5ad file including all integration results
├── project_name_raw_added.h5ad # This h5ad also contains raw UMI counts
...
The project_name.h5ad file can be updated to Cellxgene VIP platform for visualization (See section 6).
A full description of output files can be seen here.
The DE analysis in this pipeline is designed to compare “alt” and “ref” cells from “group” within each entry of “cluster” considering “sample” variations. The “group” variable should contain conditions to compare, such as Mutant v.s. Control. Thus, this pipeline is designed to loop through each cluster, and perform DE analysis between “alt” v.s. “ref”.
We strongly suggest that the reference-based label transfer has been run before DE analysis, so we can run DE based on meaningful cell types, rather than the cluster numbers automatically assigned by the pipeline.
Here, we demo the DE analysis using one real dataset. The screenshot displays the Cellxgene VIP platform:

In the first example, we would like to run DE analysis between ‘Female’ and ‘Male’ for each cluster annotated by predicted.celltype1(Created by reference-based label transfer, setting ref_name in the config.yml file). The first column assigns a name to the current comparison. In the second column, we input the header name library_id, which annotates the data sources. Then we add predicted.celltype1 in the cluster column, which allows the pipeline to loop through each cluster in predicted.celltype1. The group column contains the header name storing the comparison groups, and here we use the Sex annotation. Each time, the pipeline can only compare two conditions, such as ‘Female’ and ‘Male’. If the group column contains more groups, please list them as multiple lines in the DEGinfo.csv file.
We can also add covariates if needed, but this is optional.
Here is the DEGinfo.csv we described above:
comparisonName,sample,cluster,group,alt,ref,covars[+ separated],method[default NEBULA],model[default HL]
Compare_Female_vs_Male,library_id,predicted.celltype1,Sex,Female,Male,,NEBULA,HL
After preparing this DEGinfo.csv file, we can simply rerun the pipeline (previous steps before DE will be skipped):
scAnalyzer ~/Working_dir/config.yml
Please refer to the full tutorial for more details related to DE analysis.
scRNASequest pipeline generates an h5ad file that is fully compatible with Cellxgene VIP for data analysis and visualization.
We processed an example dataset here:
https://apps.bxgenomics.com/scrnaview/app/core/app_project_launcher.php?ID=422
You can 'Sign in as Guest' and navigate this demo data. First, make sure you select the optimal embedding using the button at the botton of the plot. We recommend 'Liger_umap' as the embedding for visualization. We can also see that categorical variables are shown on the left, while numeric variables are listed on the right. You can use the drop-shape button to highlight each variable on the plot.
The Cellxgene VIP window is close to the 'Cellxgene' logo on the top left corner. You can maximize the Cellxgene VIP window to use its functions. The annotation_1 and annotation_1_print are the labels provided by the author, see setting here. The libary_id indicates data names. The following liger_cluster and liger_cluster_predicted.celltype1 show default cluster assignment and cell type label transfer results. The last item sex shows the sex information acquired from the sample meta file.
Please refer to the GitHub website and Online tutorial for more details related to the Cellxgene VIP platform.
Please refer to this tutorial to manage and publish the project using CellDepot: https://interactivereport.github.io/CellDepot/bookdown/docs/.