This is a wrapper for competitive reads mapping (see instrain website for what is competative mapping here: https://instrain.readthedocs.io/en/master/important_concepts.html) against a collection of genomes or metagenomic assembled genomes (MAGs). And then calculate justified TAD80 values based on previous method (Rodriguez-R et.al.,2020). Bam files will be filtered first according to reads coverage and identity using CoverM. Several reads mapping softwares are supported, including bowtie2 (reads must be fasta format, gzipped or not), bwa-mem, bwa-mem2 (https://ieeexplore.ieee.org/abstract/document/8820962?casa_token=KrlJpG5fVt8AAAAA:NUlxBO2400z4M-sFMCbDn2tSXTZj_y0si_MQgNbDvPd3y223cpV-si6b8DDWCWhl-1iSI3Gh), minimap2 and bbmap. By default bbmap is used because it is faster comparing to other softwares without sacrificing accuracy (down to 70% identity, bloom filter is used to accelerate kmer matching to filter reads, this is faster). Bowtie2 --very-sensitive can also map reads down to 70% identity but parallel is not well implemented (core dumped for more than 1 thread). bwa-mem2 is 2x speedup comparing to bwa-mem with exactly the same output. Thanks to bioinformatics team at Georgia Tech (https://www.cc.gatech.edu/~saluru/)! Other mapping tools such as minimap2 only map reads down to identity 85%.
git clone https://github.com/jianshu93/Competitive_mapping
cd Competitive_mapping
chmod a+x dependencies/*
chmod a+x ./*.bash
wget http://rothlab.com/Data/T4AerOil_sbsmpl5.fa.gz
mv T4AerOil_sbsmpl5.fa.gz ./demo_input
gunzip ./demo_input/T4AerOil_sbsmpl5.fa.gz
### 1.competetive reads mapping
./compet_map_bam.bash -d ./demo_input/MAG -i ./demo_input/T4AerOil_sbsmpl5.fa -T 24 -o ./bam_out -m bbmap
### 3.calculation of justified TAD80 using coverm filter (for filtering only)
./jTAD80_new.bash -d ./bam_out -o output.txt -p 24 -c 0.75 -i 0.95 -j 0.8
All dependencies can be installed using conda. You may need to install gnu-coreutils, gawk, ggrep, gsed et.al. first via brew:
brew install coreutils
brew install gawk
brew install grep
brew install gsed
brew install brewsci/bio/bwa-mem2
git clone https://github.com/jianshu93/Competitive_mapping
cd Competitive_mapping
chmod a+x dependencies/*
chmod a+x ./*.bash
gunzip ./demo_input/T4AerOil_sbsmpl5.fa.gz
### 1.competetive reads mapping
./compet_map_bam_darwin.bash -d ./demo_input/MAG -i ./demo_input/T4AerOil_sbsmpl5.fa -T 8 -o ./bam_out -m bbmap
### 3.calculation of justified TAD80 using coverm filter (for filtering only)
./jTAD80_new_darwin.bash -d ./bam_out -o output.txt -p 4 -c 0.75 -i 0.95 -j 0.8
- In output directory you will have sorted bam files for each genome and also a big sorted bam file for all genomes. Those bam files for each genome can be used for recuitment plot (https://github.com/KGerhardt/RecruitPlotEasy) (bwa-mem and bowtie2 is supported, see an example below). The big bam file can be used for calculation of coverage depth et.al. using CoverM (https://github.com/wwood/CoverM)
- Justified TAD80 is based on BedGraph.tad.rb in enveomics (https://github.com/lmrodriguezr/enveomics) but mapped reads are filtered using CoverM before calculating.
- The filtered bam files in step 2 in the output directory can be directly used for variant calling. Softwares such as freebayes (https://github.com/freebayes/freebayes) and GATK (https://github.com/broadinstitute/gatk) can be used. See below.
bwa/bwa-mem2 mapping, MAG001, recruitment plot
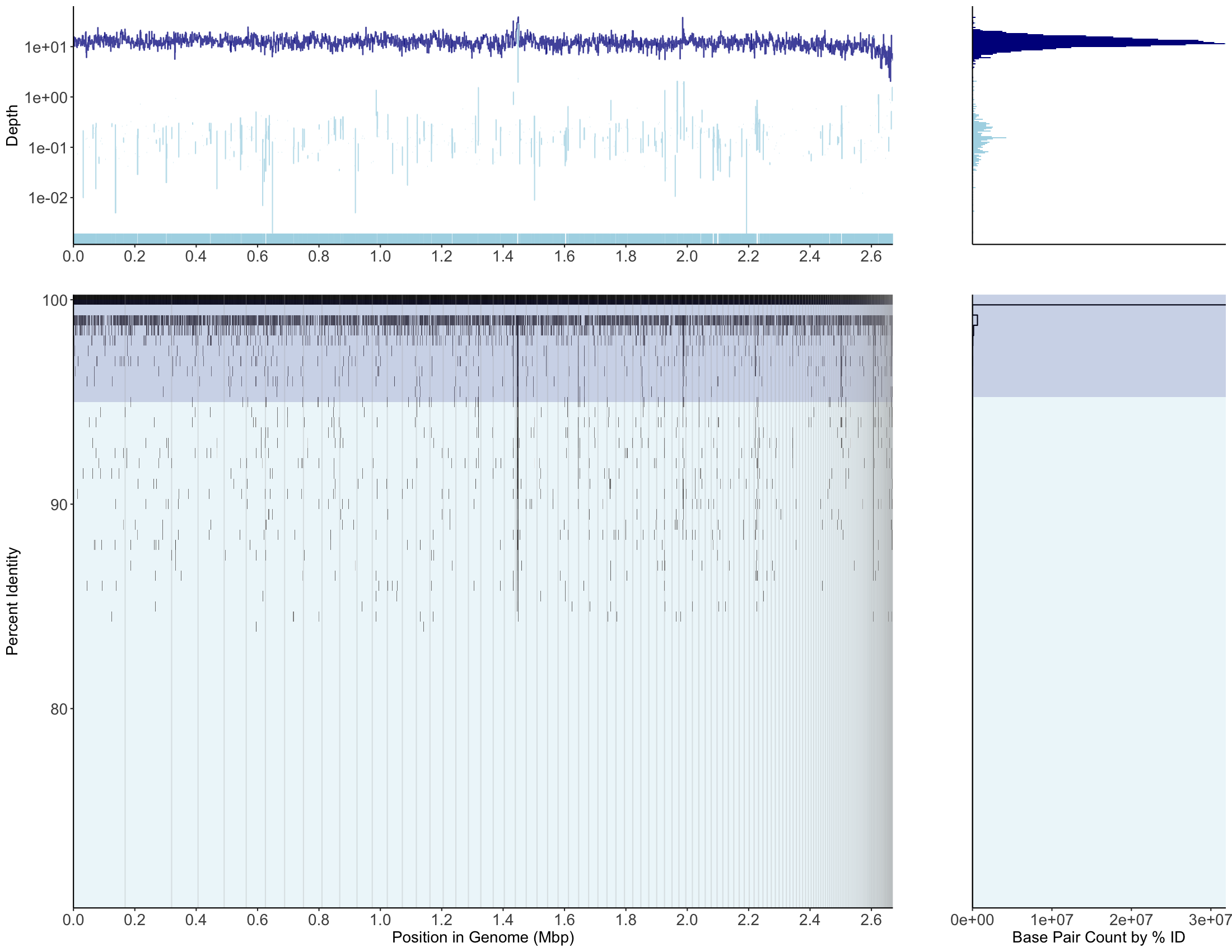
bowtie2 mapping, MAG001, recruitment plot

minimap2 mapping, MAG001, recruitment plot
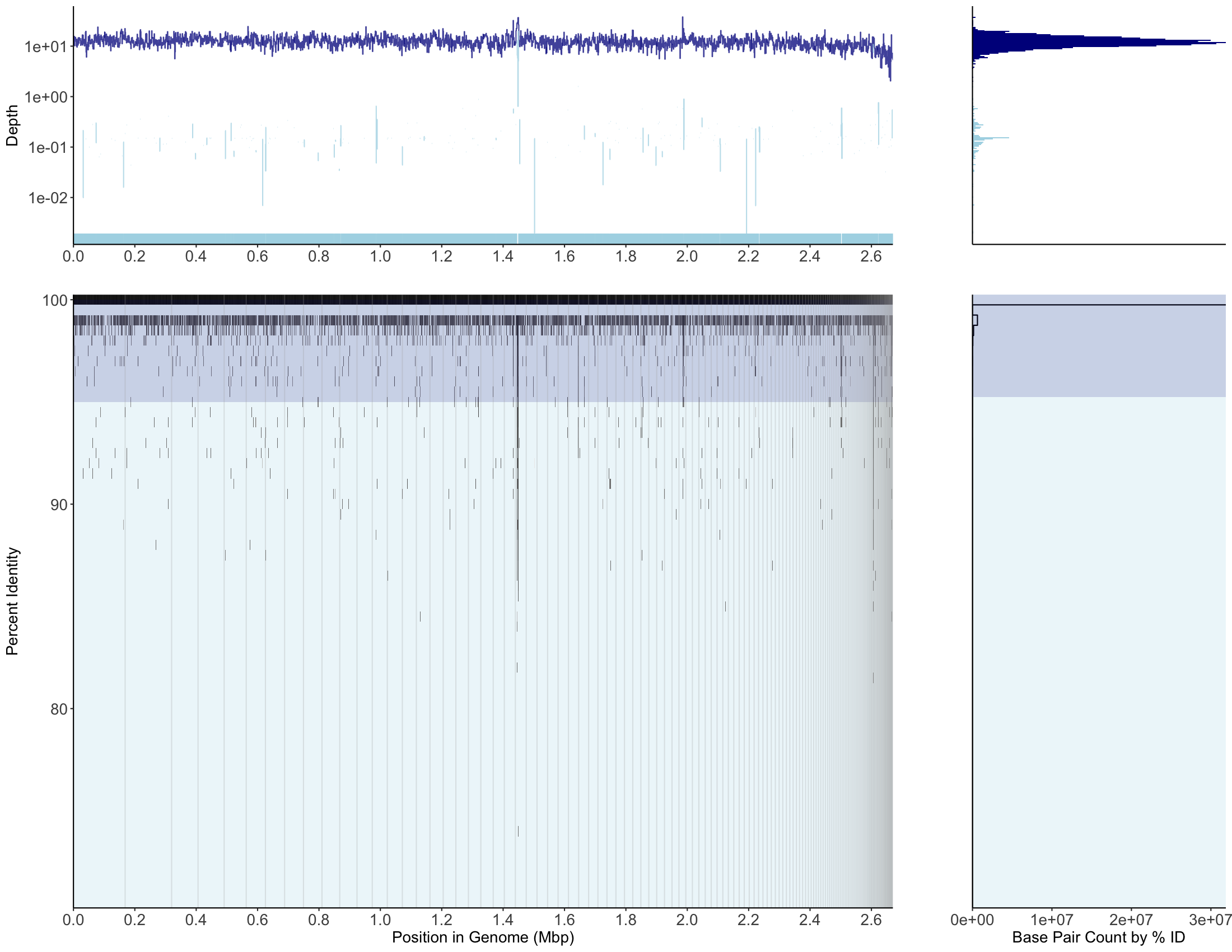
bbmap mapping, MAG001, recruitment plot
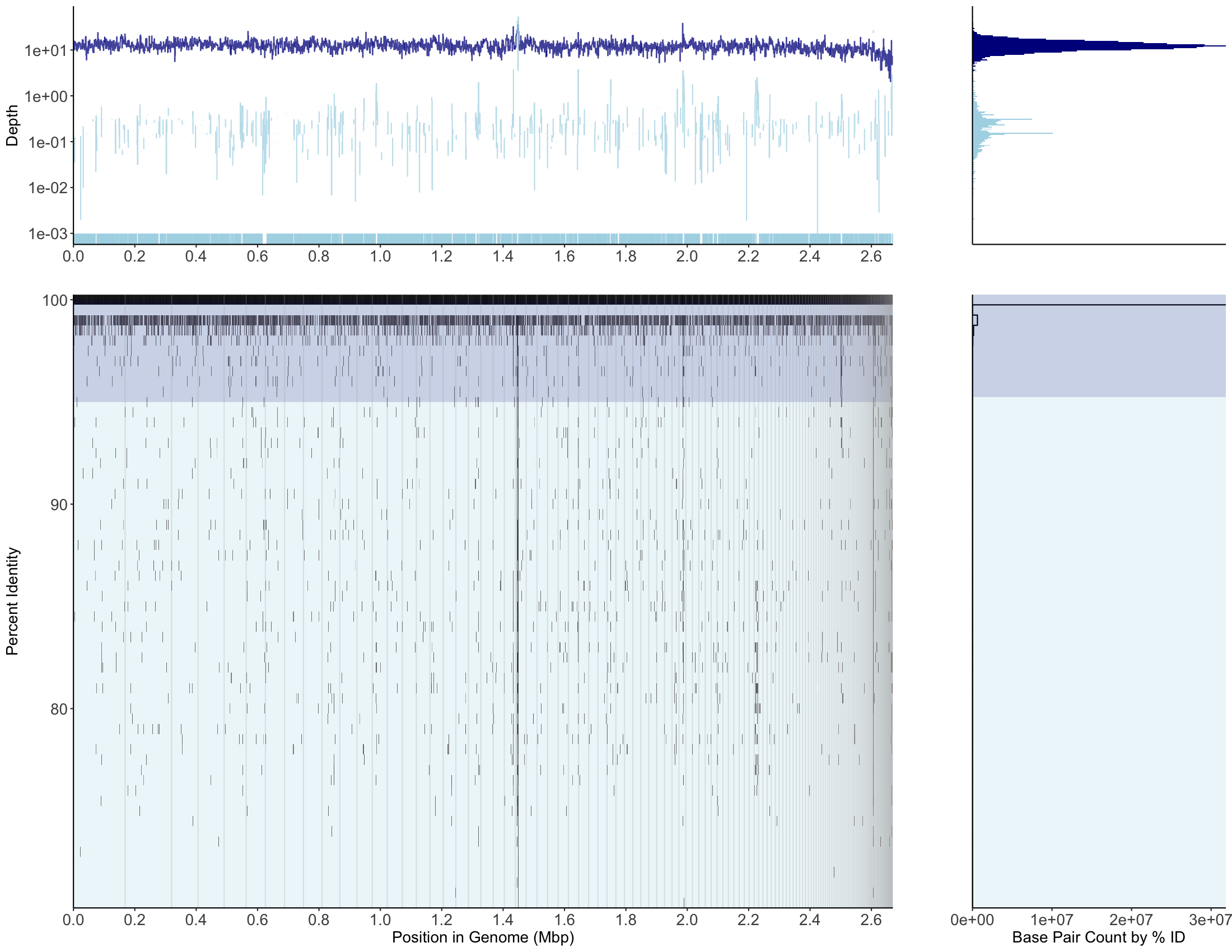
bwa/bwa-mem2
| genome_name | jTAD80 |
|---|---|
| lab5_MAG.001 | 11.983417312703843 |
| lab5_MAG.002 | 5.808299064413597 |
bowtie2
| genome_name | jTAD80 |
|---|---|
| lab5_MAG.001 | 11.914096173780646 |
| lab5_MAG.002 | 5.760955878391725 |
minimap2
| genome_name | jTAD80 |
|---|---|
| lab5_MAG.001 | 11.981560901976831 |
| lab5_MAG.002 | 5.802688239625047 |
bbmap
| genome_name | jTAD80 |
|---|---|
| lab5_MAG.001 | 11.97209484596707 |
| lab5_MAG.002 | 5.754495663260725 |
freebayes -f ./bam_out/lab5_MAG.001.fasta ./bam_out/lab5_MAG.001.filtered.sorted.bam > lab5_MAG.001.vcf
Then population genomic analysis such as dN/dS ratio and genomic diversity pi can be calculated using POGENOM (https://github.com/EnvGen/POGENOM)
Sample data from Karthikeyan et.al.,2021, Env.Sci.Tech. After running the mapping script you will have a bam_out directory.
### for Linux
cd Competitive_mapping
./dependencies/coverm_linux genome -d ./bam_out/ -x fasta -b ./bam_out/all_mags_rename_sorted.bam -m trimmed_mean --trim-min 0.1 --trim-max 0.9 --min-read-percent-identity 0.95 --min-read-aligned-percent 0.75
### for MacOS
cd Competitive_mapping
./dependencies/coverm_darwin genome -d ./bam_out/ -x fasta -b ./bam_out/all_mags_rename_sorted.bam -m trimmed_mean --trim-min 0.1 --trim-max 0.9 --min-read-percent-identity 0.95 --min-read-aligned-percent 0.75
| genome_name | jTAD80 (bwa-mem2) | Trimmed_Mean80 (coverm genome, bwa-mem2) | jTAD80 (bbmap) | jTAD80 (bowtie2) | jTAD80 (minimap2) |
|---|---|---|---|---|---|
| MaxBin.001 | 237.6306129 | 237.69215 | 237.48718380617993 | 237.08225972882352 | 236.9699149892184 |
| MaxBin.012 | 102.6480596 | 102.73 | 101.83691770829172 | 102.15607345234928 | 102.24843144795943 |
| MaxBin.035_sub | 6.394396129 | 6.6497817 | 6.359137268165782 | 6.372328363069162 | 6.366147391600805 |
| MaxBin.047 | 22.70832466 | 22.750353 | 22.515260517709756 | 22.60274518449622 | 22.559893374930745 |
| MaxBin.051 | 12.87198109 | 13.105129 | 12.742882817890578 | 12.799255459939497 | 12.778210263442002 |
| MetaBAT.009 | 9.244269553 | 9.455999 | 9.101152594306223 | 9.17924063788168 | 9.165697952209916 |
| MetaBAT.015 | 20.03221762 | 20.185205 | 19.41191468220363 | 19.85043892169667 | 19.836915591788394 |
| MetaBAT.016 | 25.42493589 | 25.41395 | 25.350523707300205 | 25.353831995850683 | 25.324779567491248 |
| MetaBAT.017 | 11.62968809 | 11.843799 | 11.426674323695218 | 11.567241963234936 | 11.521406831745859 |
| MetaBAT.019 | 12.98588635 | 13.086388 | 12.960811281625881 | 12.958032351721277 | 12.940247200331813 |
| MetaBAT.024 | 24.59416783 | 24.556606 | 24.322561240202077 | 24.457514269667325 | 24.457448599446483 |
| MetaBAT.026 | 23.39601346 | 23.39584 | 23.159304411069343 | 23.278743959361996 | 23.2904193498208 |
| MetaBAT.029 | 11.71779875 | 11.882411 | 11.589002826858984 | 11.685213871085764 | 11.600488192903546 |
| MetaBAT.030 | 9.325849312 | 9.539687 | 9.25759262387727 | 9.29696657053332 | 9.260323776310972 |
bwa, seqtk, CoverM, ruby, samtools, bedtools and minimap2 are required for this pipeline. freebayes can also be installed via conda
### bwa-mem2 is only supported for linux in conda channel but you can installed on MacOS via brew
conda install -c bioconda bwa freebayes samtools bedtools seqtk minimap2 bwa-mem2
CoverM can be installed here:https://github.com/wwood/CoverM
I contributed to v0.5.0 for CoverM on comparing bedtools -genomecov and samtools depth to coverm genome, coverm does not take care of secondary alignemnts at the first place but both samtools and bedtools does. It was fixed in the v0.5.0. See here: https://github.com/wwood/CoverM/releases/tag/v0.5.0
Li, H and R Durbin. 2009. “Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform.” 25(14):1754–60.
Li, Heng et al. 2009. “The Sequence Alignment/Map Format and SAMtools.” Bioinformatics 25(16):2078–79.
Quinlan, Aaron R. and Ira M. Hall. 2010. “BEDTools: a Flexible Suite of Utilities for Comparing Genomic Features.” Bioinformatics 26(6):841–42.
Garrison, Erik and Gabor Marth. 2012. “Haplotype-Based Variant Detection From Short-Read Sequencing.” 1–9. Retrieved (https://arxiv.org/abs/1207.3907).
Li, Heng et al. 2009. “The Sequence Alignment/Map Format and SAMtools.” Bioinformatics 1–2.
Rodriguez-R, L M., D Tsementzi, C Luo, and K T. Konstantinidis. 2020. “Iterative Subtractive Binning of Freshwater Chronoseries Metagenomes Identifies Over 400 Novel Species and Their Ecologic Preferences.” Environmental Microbiology 1462–2920.15112–67.
Smruthi Karthikeyan, Minjae K. P. H.-R. J. K. H. J. C. S. W. A. O. M. H. J. E. K. A. K. T. K. 2020. “Integrated Omics Elucidate the Mechanisms Driving the Rapid Biodegradation of Deepwater Horizon Oil in Intertidal Sediments Undergoing Oxic−Anoxic Cycles.” Environmental Science & Technology 54(16):1–12.
Li, Heng. 2013. “Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM.” arXiv 1–3.
Vasimuddin, Md, Sanchit Misra, Heng Li, and Srinivas Aluru. 2019. “Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems.” IEEE International Parallel and Distributed Processing Symposium 314–24.
Subramaniyan, Arun et al. 2020. “Accelerating Maximal-Exact-Match Seeding with Enumerated Radix Trees.” arXiv 1–10.
Li, Heng. 2018. “Minimap2: Pairwise Alignment for Nucleotide Sequences.” Bioinformatics 1–7.
Ben Langmead, Christopher Wilks, and Rone Charles. 2019. “Scaling Read Aligners to Hundreds of Threads on General-Purpose Processors.” Bioinformatics 1–12.
Brian Bushnell, 2018, https://github.com/BioInfoTools/BBMap