BCAW Tool manages a complete automated workflow to analyze the codon usage bias for genes and genomes of any organism. For thus who can not use python, we provide here a GUI software that can work on any operating system, with the minimal requirement.
- Install Python >=3.7.
- Download BCAWT using this command
pip install BCAWT- Download this zip file. After, compress the file and run BCAWT-GUI.py script by any way you would like.
1- Run the "BCAWT-GUI.py"
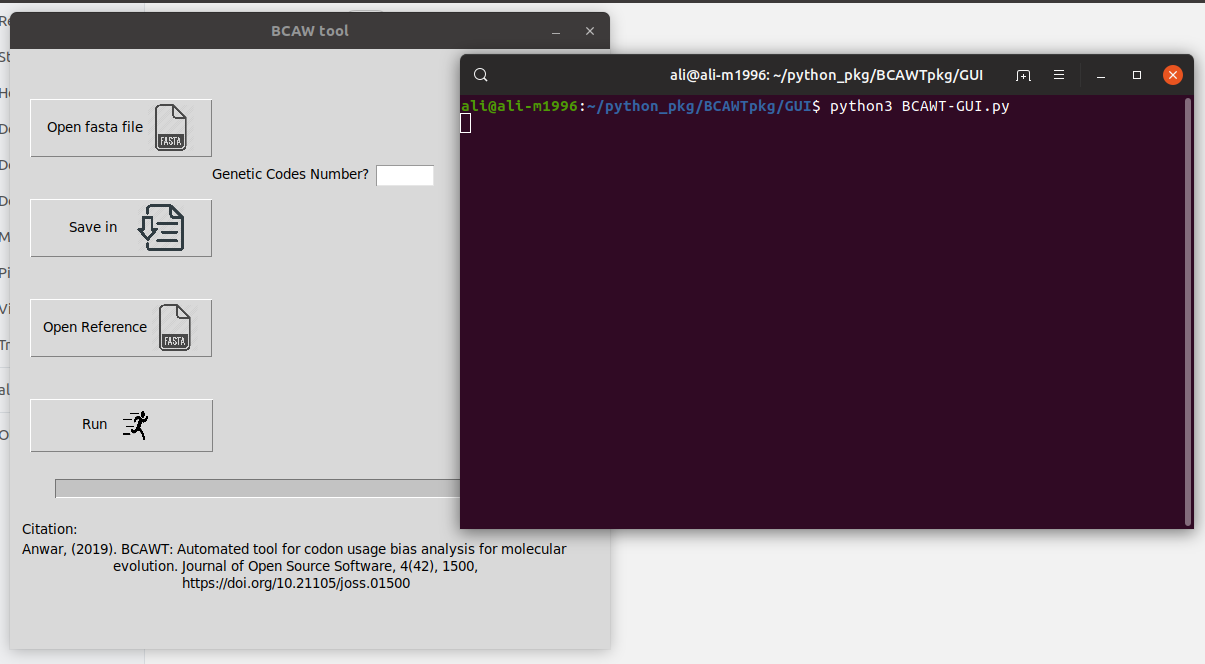
- Import a fasta file contains the genes you want to test (from "Open fasta file" button).
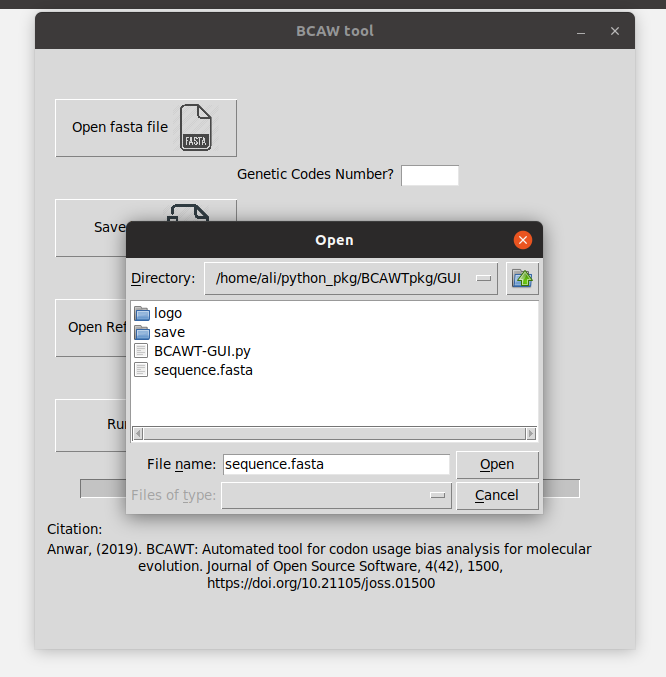
- Enter the genetic code your genes use (for more information see: NCBI)
- Choose a directory (folder) you want to save the result in (from "Save in" button).
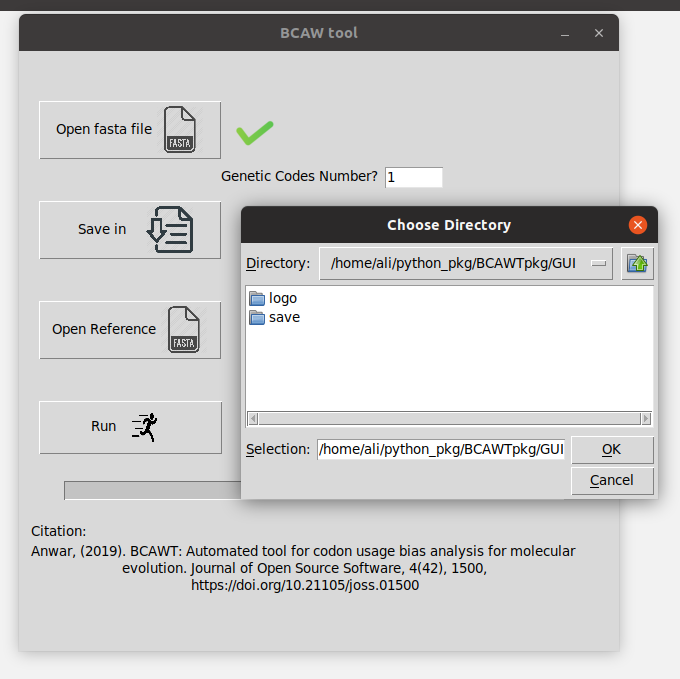
- Import a fasta file contains genes that will be used as a reference set (or just do not use it if you want BCAWT to generate the reference set automatic, for more information see: BCAWT) (from "Open Reference" button).
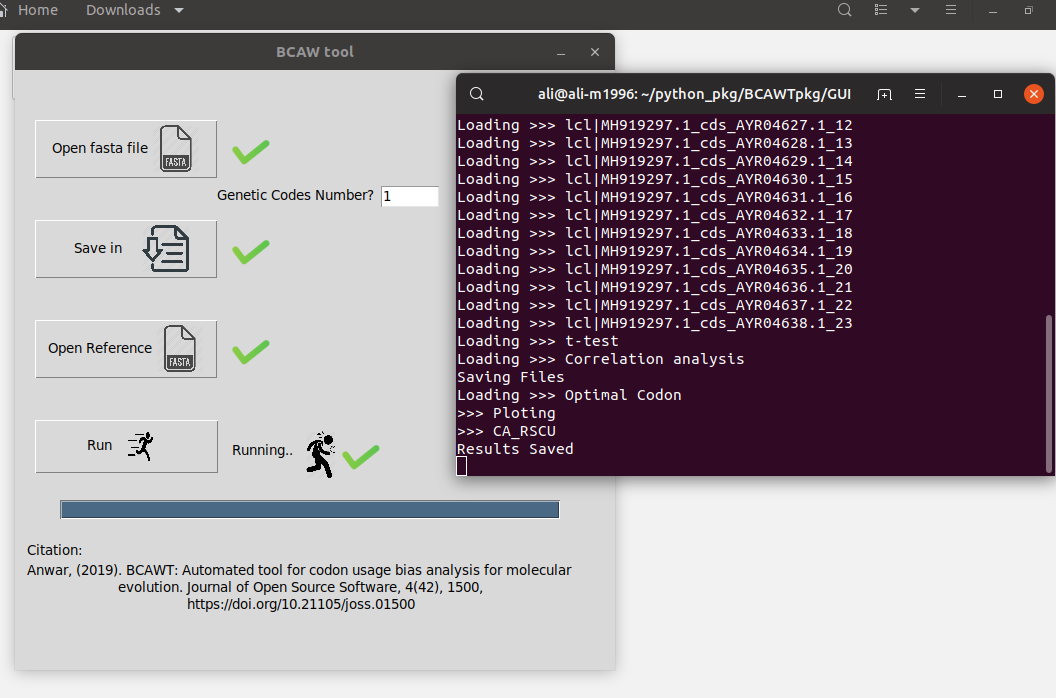
- Now, just Run it. Wait for the loading bar to finish.
Contributions to the software are welcome
For bugs and suggestions, the most effective way is by raising an issue on the github issue tracker. Github allows you to classify your issues so that we know if it is a bug report, feature request or feedback to the authors.
Anwar, (2019). BCAWT: Automated tool for codon usage bias analysis for molecular evolution. Journal of Open Source Software, 4(42), 1500, https://doi.org/10.21105/joss.01500