Tony Yang (tony@tony.tc)
Mentored by Andy Yates and Leanne Haggerty from EBI Ensembl
In this GSoC project, three phased goals were achieved.
- A program to calculate the percentage of guanine and cytosine in genomic sequences. (repository link)
- Common Workflow Language (cwl) workflows that perform genome analysis. (workflows, commandline tools)
- A framework for running workflows on newly reported genome assemblies automatically. (scripts)
With the development of new sequencing machinery and their much-reduced cost, a large quantity of novel genome data is produced daily at an increasing rate by the government and private funded genome projects. To accelerate genomic data analysis and gene annotation, it is crucial to process those primary data as they are produced automatically.
GC content, the percentage of guanine (G) or cytosine (C) bases on a genome, provides vital information about this organism. Guanine (G) and cytosine (C) base pairs stack thermodynamically more favourable than adenine (A) and thymine (T) base pairs, hence stabilise the DNA. GC content is varied for different organisms and can provide evolutionary insights into particular organisms. A higher than background GC-content can indicate coding regions in a genomic sequence, which is useful to facilitate gene annotation.
A large variety of workflow languages were developed to connect tools and form pipelines but they lack portability and reproducibility. Common Workflow Language (CWL) was introduced as an open standard which serves data-intensive science. CWL documents, written in JSON or YAML, are used to describe the connection of different command line tools. Workflows described in CWL specification are easily portable and scalable in different computational environments, hence it is ideal for this project.
I started by writing a GC program in Python, which was the goal of the first phase of this GSoC project. This commandline program would take a fasta formatted file which contains one or more genomic sequences, and calculate the GC percentage with a specified window-szie and shift then write the result to file. The output file format can be chosen among wiggle file, gzip compressed wiggle file or bigwig file. The results of multi-sequence input file can be a single file or separated to one sequence per file. This GC program can be executed as a Python script, from the packaged binary file, install the package from PYPI or run the docker image as a container. For detailed usage and options, please see the repository.
For the second stage, I containerised my GC program into a docker image, along with several other tools and scripts. I also developed CWL commandline scripts for each of the tools (see tools) and workflows to run all the analyses by providing an ENA accession number (see workflows). These CWL files set the stage for the deployment of the workflow to clusters.
Third-party tools included in the workflows are:
-
Tandem Repeat Finder from Boston University:
G. Benson, "Tandem repeats finder: a program to analyze DNA sequences" Nucleic Acids Research (1999) Vol. 27, No. 2, pp. 573-580.
Link -
CpG island from UCSC:
Description
Source
For the final stage, I deployed the workflows to EBI's cluster. My Python scripts would retrieve a list of assemblies from an internal database, which is updated daily, consists of the ENA accessions for all the assemblies on ENA's archive. Subsequently, my scripts update_tables.py would update my own MySQL database which holds information about the assemblies and their chromosomes, together with all jobs and their statuses. submit_jobs.py will generate the YAML input files for CWL and submit all waiting jobs to LSF with Toil (a CWL executor with batch system support). Finally, process_result.py would parse the CWL output and update the jobs' statuses accordingly, then aggregate the result to determine if a whole assembly's jobs are all done.
Here are the graphs of the two worlflows visualised with CWL viewer
Fasta_GC_TRF_CpG.png
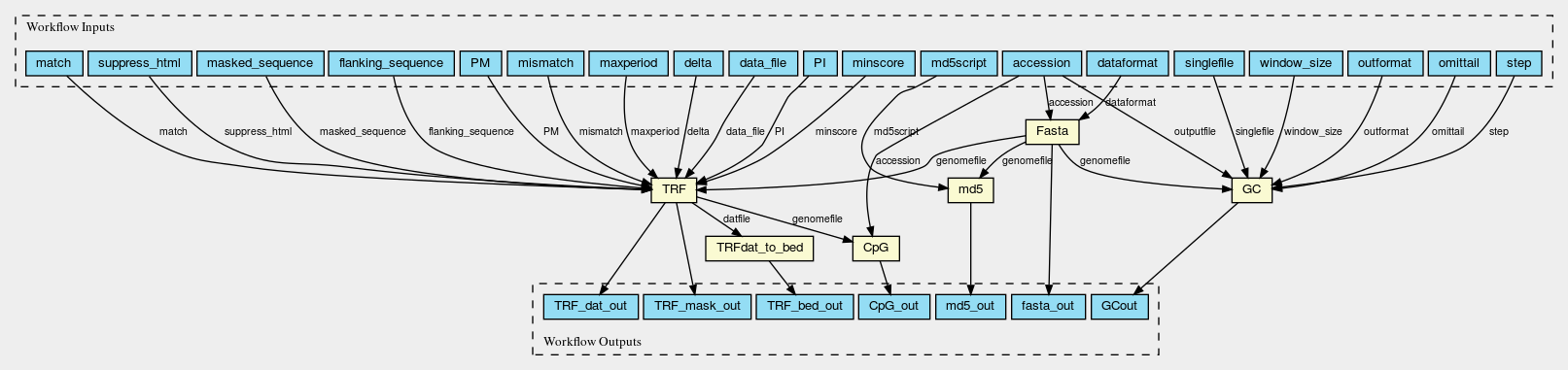 Permalink
Permalink
Gz_Fasta_GC_TRF_CpG.png
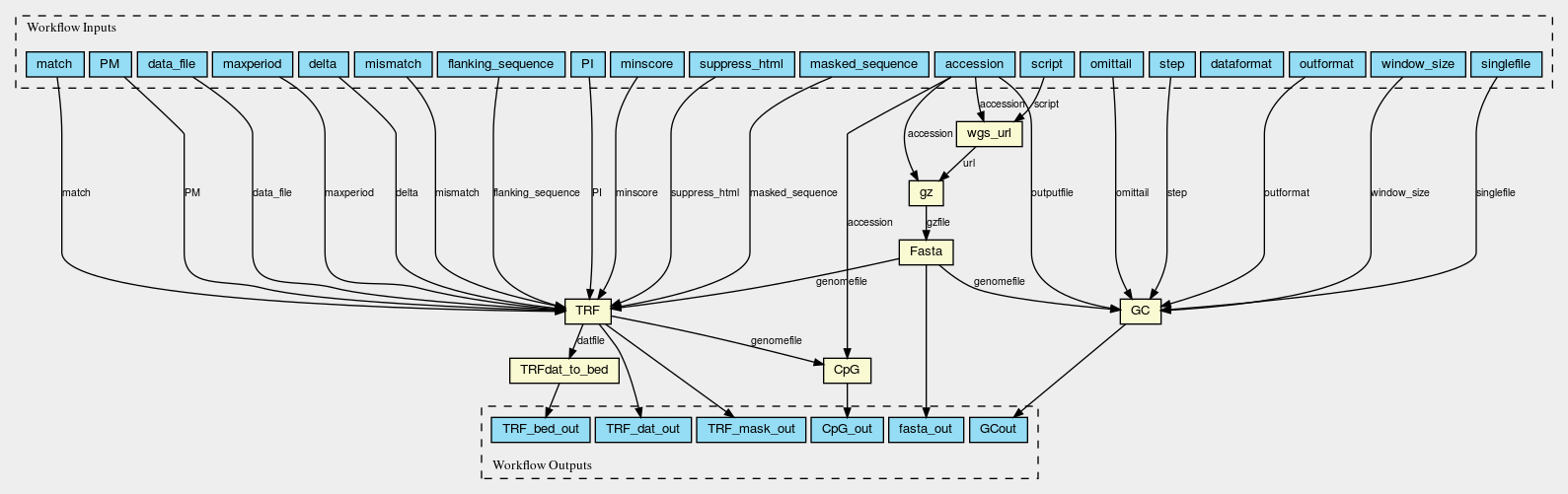 Permalink
Permalink
Ensembl has an internal registry database which stores information about all the assemblies, which looks like
mysql> select * from assembly;
| assembly_id | chain | version | stable_id_space_id | ... | ... |
|-------------|---------------|---------|--------------------|-----|-----|
| 42389 | GCA_000090745 | 1 | 1 | ... | ... |
| ... | ... | ... | ... | ... | ... |
The columns I really need are chain and version which give me the accession GCA_000090745.1 when combined. This database updates daily.
My script update_tables.py checks if the accession already presents in my GCA table. GCA table looks like
mysql> select * from GCA limit 5;
+----+-----------------+--------+---------+-----------------+
| id | accession | status | records | assembly_level |
+----+-----------------+--------+---------+-----------------+
| 1 | GCA_000090745.1 | 0 | 13 | chromosome |
| 2 | GCA_000003055.3 | 0 | 30 | chromosome |
| 3 | GCA_000002985.3 | 0 | 6 | complete genome |
| 4 | GCA_002754865.1 | 0 | 39944 | scaffold |
| 5 | GCA_000002285.2 | 0 | 39 | chromosome |
+----+-----------------+--------+---------+-----------------+
If there are new assemblies in the registry but not in my GCA table, the script will add the new accessions to my table and try to pull the xml file on this assembly. An example of the xml file contains useful information about the assembly. The number of chromosomes/scaffolds/contigs (depends on assembly level) and the assembly level are parsed to include in the table as shown above. The status column indicates if all the analyses on this assembly are finished, 0 denotes finished and unfinished ones will be left as NULL.
It is important to note that assemblies assembled at chromosome level and at scaffold/contig level have slightly different representations on the ENA website. Assemblies at chromosome level have accessions to all the chromosomes listed in the xml file. Assemblies at scaffold/contig level have all the sequences in one gzip compressed FASTA file available directly from the assembly page. This will cause different behaviors in later stages, hence two workflows are used to treat them differently.
Once the GCA table is updated, if the new entry is assembled at chromosome level, the assembly's chromosomes' accession will be added to a separate table called Chromosome, which looks like
mysql> select * from Chromosome limit 5;
+----+-----------------+------------+----------------------------------+-----------+------+--------+
| id | GCA_accession | accession | md5 | length | name | status |
+----+-----------------+------------+----------------------------------+-----------+------+--------+
| 1 | GCA_000090745.1 | CM000937.1 | 63a54646c356d245fa0fe09d8dc8e6e9 | 263920458 | 1 | 0 |
| 2 | GCA_000090745.1 | CM000938.1 | 5a19b466933609385e01040025117dca | 199619895 | 2 | 0 |
| 3 | GCA_000090745.1 | CM000939.1 | 4ae44e1aea37da6b4fd8b63b4f1d6be4 | 204416410 | 3 | 0 |
| 4 | GCA_000090745.1 | CM000940.1 | fc6cb6300a1011ddfab01e6de5dbb7b2 | 156502444 | 4 | 0 |
| 5 | GCA_000090745.1 | CM000941.1 | b7fc968a673ca477f3bd9d8afc5bc961 | 150641573 | 5 | 0 |
+----+-----------------+------------+----------------------------------+-----------+------+--------+
Once all the chromosomes of the assemblies are added to Chromosome table, the script will try to retrieve the xml record of the chromosomes, which contains the md5 checksum of the chromosome's genomic sequence and the length of the sequence. The name field of the chromosome comes from the xml record of the assembly, not individual chromosome. Again, status = 0 denotes all jobs on this chromosome are finished and status = 1 means there are unfinished jobs.
Once all the chromosomes are added to Chromosome table, each chromosome will be added to the Jobs table together with all the assemblies assembled at lower than chromosome level (scaffold and contig levels). The Jobs table looks like
mysql> select * from Jobs limit 12;
+----+----------------------+-----------+------------------------------------------+--------+
| id | chromosome_accession | job_name | SHA1 | status |
+----+----------------------+-----------+------------------------------------------+--------+
| 1 | CM000937.1 | get_fasta | f6fe4f4e9118657bf60a0b258ddde422875d3606 | 0 |
| 2 | CM000937.1 | GC | b7d370a7f327e892870a7966f1ac1025e2e2c8ac | 0 |
| 3 | CM000937.1 | trf | 4c6e1ac96c2160e28f749b0cfd3b032a3129a4a6 | 0 |
| 4 | CM000937.1 | CpG | 2b6de0ee407befaa9e532cb09b41c01cb989de02 | 0 |
| 5 | CM000938.1 | get_fasta | 5a3a544518f4aebdd1058b71c61365a3f0a31114 | 0 |
| 6 | CM000938.1 | GC | ccf4d2948e5b6881d96575a201177d2be84e7b66 | 0 |
| 7 | CM000938.1 | trf | e014826e37aca6730e60492f425c1af6b772ccbd | 0 |
| 8 | CM000938.1 | CpG | afbba61c52e9f9f8b54b37a3b70ab359529a436a | 0 |
| 9 | CM000939.1 | get_fasta | e47cdaf2f2d928a1657652a78e257cbd96e3e959 | 0 |
| 10 | CM000939.1 | GC | 474146b4334d520f7c9894ce98607f73cfe4311e | 0 |
| 11 | CM000939.1 | trf | 82d1edc88a3a1736217d688162b2aae3d26ac00e | 0 |
| 12 | CM000939.1 | CpG | 6281ef42ec82941d5161673632d331dea7a6d126 | 0 |
+----+----------------------+-----------+------------------------------------------+--------+
The default values for SHA1 and status are NULL when added. Note that chromosome_accession column contains accessions for both chromosomes and assemblies at scaffold/contig level. The job_name field contains each analysis needed for each sequence. When the jobs are submitted by submit_jobs.py, the status for the job will be set to 1. Once the job is finished, process_result.py script will check if the result files exist and parse Toil's stdout to get the SHA$1 checksum of each output file then update the status to 0.
On EBI's cluster, crontab is set to run update_tables.py every day at 1 a.m. submit_jobs.py will run twice a day and there will be maximum 350 workflows running concurrently in order to avoid Toil.leader saturate the jobs queue. process_result.py will run once a day to update tables to log finished jobs. When submit_jobs.py submits jobs, the script looks for jobs where the status is NULL, the job's status is set to 1 after submission. When process_result.py thinks the job finished correctly, the job's is set to 0 and the SHA$1 checksum is added to the table; if the job didn't finish correctly, the status will be set to NULL and waiting for a rerun.
It should be noted that two slightly different workflows are used for assemblies at scaffold/contig level and chromosomes, because the WGS_FASTA_FILE for assemblies at scaffold/contig level needs to be decompressed into FASTA file first so an additional decompression step is needed. Fasta_GC_TRF_CpG.cwl is used for chromosomes; Gz_Fasta_GC_TRF_CpG.cwl is used for scaffolds and contigs.
After checking all the finished jobs, process_result.py will back-propagate the results from Jobs table to Chromosome table and GCA table. If the sum of all the status under one accession is 0 in the Jobs table, the respective chromosome/assembly's status will be updated to 0. Upon completion, the script then looks through Chromosome table, if the sum of the status of all the chromosomes under one GCA_accession is 0, the status of the assembly will be marked as 0. When an assembly at chromosome level is finished, the folders of the chromosomes are copied to the assembly's folder.
This workflow is designed to be modular and additional analysis can be added easily. The only modification necessary other than the cwl workflow itself is to add additional job_name in Jobs table and add tests for completion in process_result.py script.
This project serves as a proof-of-concept that it is feasible to deploy a similar system for automated processing of primary genomic data. Currently, out of the 4000 accessions, 186 failed, most of the failure was due to unavailable FASTA file from ENA's archive. There are around 10 jobs hung for a long time or failed with no obvious reason. It is important and curious to look deeper into why they failed/could not complete.
In the long term, more analyses can be included in the workflow to provide more comprehensive data. In addition, we are looking into how to provide easy and convenient public access to the analysis results.
I would like to thank Andy Yates and Leanne Haggerty for taking the time to mentor me and providing invaluable suggestions. I truly appreciate their constant trust and encouragement.