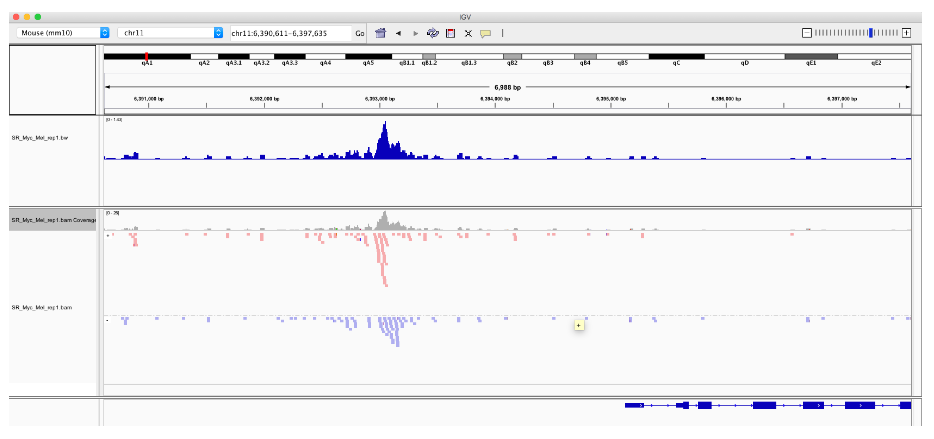
现在我们有了 BAM 文件的索引,我们可以使用 idxstatsBam() 函数检索和绘制映射读取的数量。
mappedReads <- idxstatsBam("SR_Myc_Mel_rep1.bam")
TotalMapped <- sum(mappedReads[, "mapped"])
ggplot(mappedReads, aes(x = seqnames, y = mapped)) + geom_bar(stat = "identity") +
coord_flip()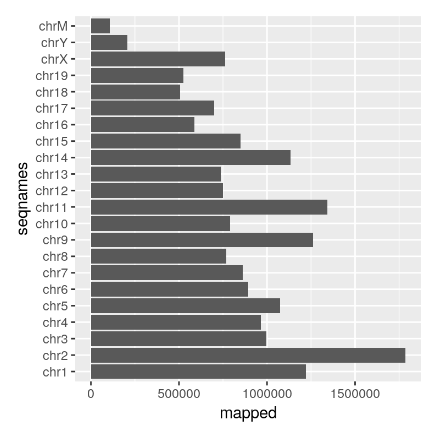
我们还可以从我们排序的、索引的 BAM 文件中创建一个 bigWig,以允许我们快速查看 IGV 中的数据。
首先,我们使用 coverage() 函数创建一个包含我们的覆盖率分数的 RLElist 对象。
forBigWig <- coverage("SR_Myc_Mel_rep1.bam")
forBigWig我们现在可以使用 rtracklayer 包的 export.bw() 函数将 RLElist 对象导出为 bigWig。
library(rtracklayer)
export.bw(forBigWig, con = "SR_Myc_Mel_rep1.bw")我们可能希望标准化我们的覆盖范围,以便我们能够比较样本之间的富集。
我们可以使用 coverage() 中的权重参数将我们的读取缩放到映射读取数乘以一百万(每百万读取数)。
forBigWig <- coverage("SR_Myc_Mel_rep1.bam", weight = (10^6)/TotalMapped)
forBigWig
export.bw(forBigWig, con = "SR_Myc_Mel_rep1_weighted.bw")