首先,我们需要将来自 MACS2 的峰值调用读取到 R 中。
我们将审查的 Myc peak 调用位于 peaks 目录中,因此我们在这里使用 dir() 函数列出与我们预期的文件模式匹配的所有文件。
peakFiles <- dir("data/peaks/", pattern = "*.peaks", full.names = TRUE)
peakFiles
我们可以循环遍历我们的制表符分隔文件(伪装成 .xls 函数)并使用循环将它们作为 data.frames 列表导入到 R 中。
macsPeaks_DF <- list()
for (i in 1:length(peakFiles)) {
macsPeaks_DF[[i]] <- read.delim(peakFiles[i], comment.char = "#")
}
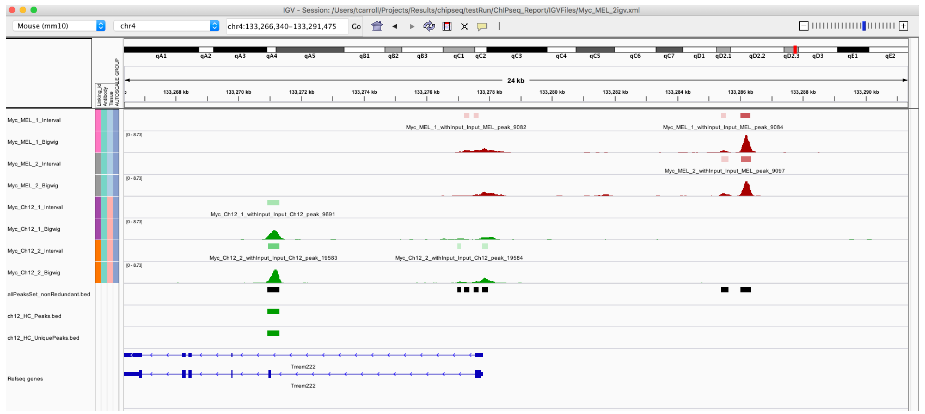
length(macsPeaks_DF)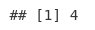
现在有了我们的 data.frames 峰值调用列表,我们循环遍历列表并为每个峰值调用创建一个 GRanges。
请记住,您也可以使用 rtracklayer 的导入功能来执行此操作。
library(GenomicRanges)
macsPeaks_GR <- list()
for (i in 1:length(macsPeaks_DF)) {
peakDFtemp <- macsPeaks_DF[[i]]
macsPeaks_GR[[i]] <- GRanges(seqnames = peakDFtemp[, "chr"], IRanges(peakDFtemp[,
"start"], peakDFtemp[, "end"]))
}
macsPeaks_GR[[1]]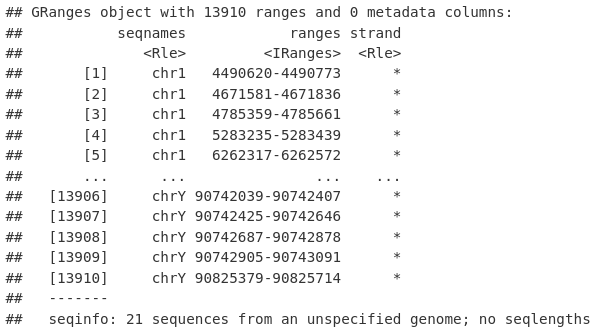
我们将要为我们的峰值呼叫分配一组合理的名称。
我们可以将 gsub() 和 basename() 函数与我们的文件名一起使用来创建一些样本名称。
basename() 函数接受文件路径(例如我们的 bam 文件的路径)并仅返回文件名(删除目录路径)。
gsub() 函数接受要替换的文本、替换文本和要替换的字符向量。
fileNames <- basename(peakFiles)
fileNames
sampleNames <- gsub("_peaks.xls", "", fileNames)
sampleNames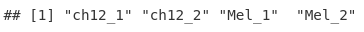
现在我们有一个作为 GRanges 对象的峰值调用的命名列表。
我们可以使用 GRangesList() 函数将 GRanges 对象列表转换为 GRangesList。
macsPeaks_GRL <- GRangesList(macsPeaks_GR)
names(macsPeaks_GRL) <- sampleNames
class(macsPeaks_GRL)
names(macsPeaks_GRL)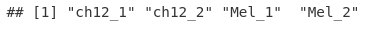
GRangesList 对象的行为与我们的标准列表一样。在这里,我们使用 lengths() 函数来获取每个重复中的峰数。
lengths(macsPeaks_GRL)
GRangesList 对象的一个主要优点是我们可以将许多 GRanges 访问器和运算符函数直接应用于我们的 GRangesList。
这意味着如果我们希望通过通用方法更改我们的 GRanges,则无需应用并转换回 GRangesList。
library(rtracklayer)
macsPeaks_GRLCentred <- resize(macsPeaks_GRL, 10, fix = "center")
width(macsPeaks_GRLCentred)
现在我们有了 GRangesList,我们可以提取 Mel 复制的峰值调用。
Mel_1_Peaks <- macsPeaks_GRL$Mel_1
Mel_2_Peaks <- macsPeaks_GRL$Mel_2
length(Mel_1_Peaks) # ## [1] 13777
length(Mel_2_Peaks) # ## [1] 13512我们可以使用 %over% 运算符提取唯一的峰值调用以复制 1 或 2。
Mel_1_Unique <- Mel_1_Peaks[!Mel_1_Peaks %over% Mel_2_Peaks]
Mel_2_Unique <- Mel_2_Peaks[!Mel_2_Peaks %over% Mel_1_Peaks]
length(Mel_1_Unique) # ## [1] 4668
length(Mel_2_Unique) # ## [1] 4263
export.bed(Mel_1_Unique, "Mel_1_Unique.bed")
export.bed(Mel_2_Unique, "Mel_2_Unique.bed")
同样,我们可以提取复制 1 或 2 常见的峰值调用。
然而,共同的数字不同。这是因为一个样本中的 2 个峰调用可以与另一个重复中的 1 个峰调用重叠。
Mel_1_Common <- Mel_1_Peaks[Mel_1_Peaks %over% Mel_2_Peaks]
Mel_2_Common <- Mel_2_Peaks[Mel_2_Peaks %over% Mel_1_Peaks]
length(Mel_1_Common) # ## [1] 9109
length(Mel_2_Common) # ## [1] 9249
export.bed(Mel_1_Common, "Mel_1_Common.bed")
export.bed(Mel_2_Common, "Mel_2_Common.bed")
尽管重叠,但这些峰并不相同。那么我们如何确定几个样本的共同共识峰。

为了解决这个问题,ChIPseq 中的一个常见操作是在所有样本中定义一组非冗余峰。
为此,我们首先将所有重复的所有峰(这里是 Mel 和 Ch12)汇集到一组冗余的重叠峰中。
allPeaksSet_Overlapping <- unlist(macsPeaks_GRL)
allPeaksSet_Overlapping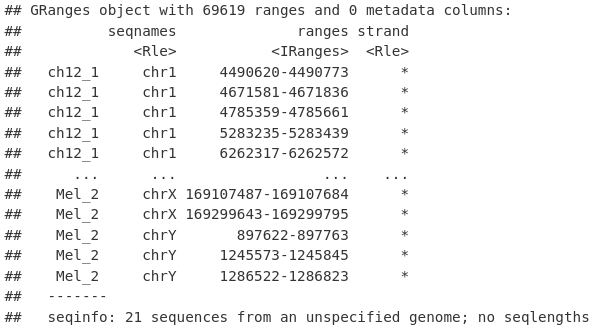
然后我们可以使用 reduce() 函数将我们的峰折叠成非冗余的、不同的峰,代表任何样本中存在的峰。
allPeaksSet_nR <- reduce(allPeaksSet_Overlapping)
allPeaksSet_nR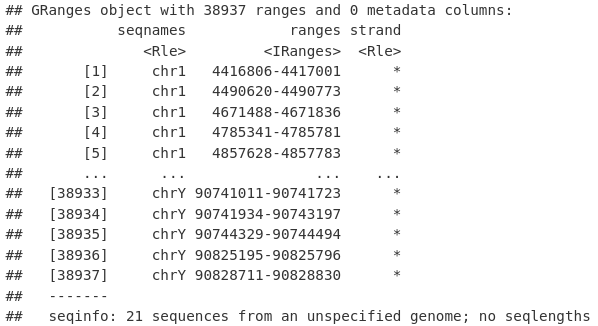
export.bed(allPeaksSet_nR, "allPeaksSet_nR.bed")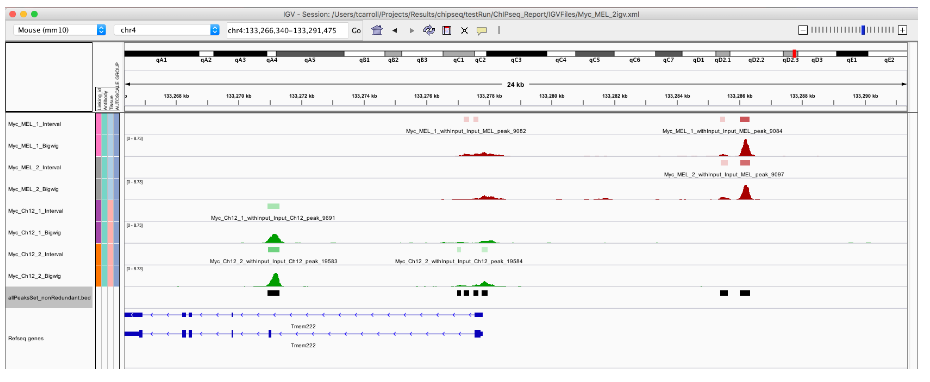
使用我们新定义的非冗余峰集,我们现在可以使用 %over% 运算符和逻辑表达式从该集中识别我们的重复中存在哪些峰。
commonPeaks <- allPeaksSet_nR[allPeaksSet_nR %over% Mel_1_Peaks & allPeaksSet_nR %over%
Mel_2_Peaks]
commonPeaks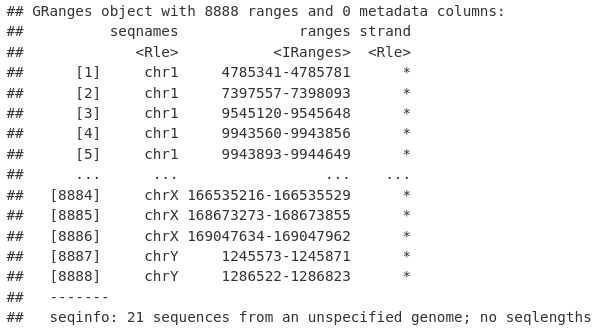
export.bed(commonPeaks, "commonPeaks.bed")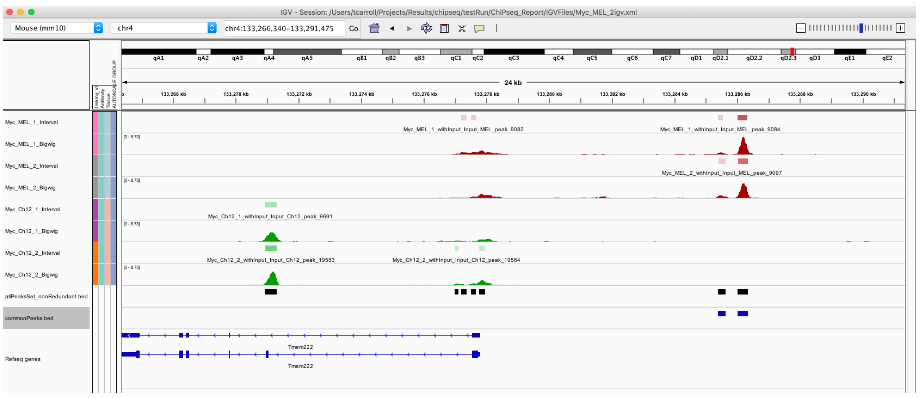
同样,我们可以确定哪些峰仅出现在一次重复中。
mel1_Only <- allPeaksSet_nR[allPeaksSet_nR %over% Mel_1_Peaks & !allPeaksSet_nR %over%
Mel_2_Peaks]
mel2_Only <- allPeaksSet_nR[!allPeaksSet_nR %over% Mel_1_Peaks & allPeaksSet_nR %over%
Mel_2_Peaks]
length(mel1_Only) # ## [1] 4445
length(mel2_Only) # ## [1] 4185
export.bed(mel1_Only, "mel1_Only.bed")
export.bed(mel2_Only, "mel2_Only.bed")
当处理大量峰时,我们通常会定义一个逻辑矩阵来描述我们的非冗余峰出现在哪些样本中。
首先,我们使用循环为每个样本中出现的非冗余峰生成一个逻辑向量。
overlap <- list()
for (i in 1:length(macsPeaks_GRL)) {
overlap[[i]] <- allPeaksSet_nR %over% macsPeaks_GRL[[i]]
}
overlap[[1]][1:2]
我们现在可以使用 to do.call 和 cbind 函数将我们的重叠列表列绑定到我们的峰值出现矩阵中。
overlapMatrix <- do.call(cbind, overlap)
colnames(overlapMatrix) <- names(macsPeaks_GRL)
overlapMatrix[1:2, ]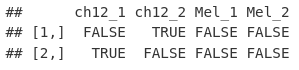
我们可以使用 mcols() 访问器将矩阵添加回非冗余峰的 GRanges() 的元数据列中。
现在我们有了非冗余峰以及每个样本中这些峰的出现,我们可以轻松识别重复和条件/细胞系特有或共有的峰。
mcols(allPeaksSet_nR) <- overlapMatrix
allPeaksSet_nR[1:2, ]
limma 包常用于分析 RNAseq 和微阵列数据,并包含许多有用的功能。
一个非常有用的函数是 vennDiagram 函数,它允许我们绘制逻辑矩阵的重叠,就像我们创建的那样。
library(limma)
vennDiagram(mcols(allPeaksSet_nR))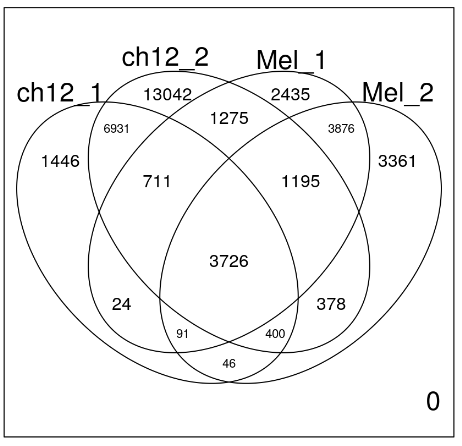
limma 包的 vennCounts 函数允许我们检索维恩图中显示的计数作为 data.frame。
vennCounts(mcols(allPeaksSet_nR))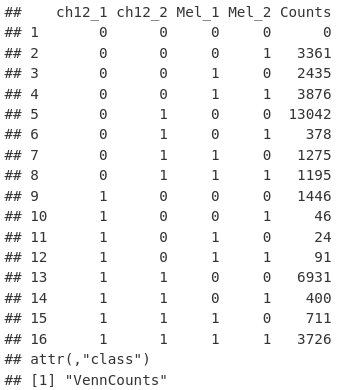
使用我们的非冗余峰集和峰出现矩阵,我们可以在条件下定义复制峰。在这里,我们定义了在两个 Ch12 重复中出现的峰值。
由于逻辑矩阵等效于 1 或 0 矩阵(1 = TRUE 和 0 = FALSE),我们可以使用 rowSums 函数在至少 2 个 Ch12 重复中提取峰。
ch12_HC_Peaks <- allPeaksSet_nR[rowSums(as.data.frame(mcols(allPeaksSet_nR)[, c("ch12_1",
"ch12_2")])) >= 2]
export.bed(ch12_HC_Peaks, "ch12_HC_Peaks.bed")
ch12_HC_Peaks[1:2, ]

同样,我们可以定义在 Ch12 中复制但在 Mel 样本中不存在的峰。
ch12_HC_UniquePeaks <- allPeaksSet_nR[rowSums(as.data.frame(mcols(allPeaksSet_nR)[,
c("ch12_1", "ch12_2")])) >= 2 & rowSums(as.data.frame(mcols(allPeaksSet_nR)[,
c("Mel_1", "Mel_2")])) == 0]
export.bed(ch12_HC_UniquePeaks, "ch12_HC_UniquePeaks.bed")
ch12_HC_UniquePeaks[1, ]