Aplikacja stworzona przy pomocy Shiny-R. Służy do przykładowej analizy danych, pochodzących z eksperymentu mikromacierzowego.
- Odczytanie plików .CEL
- Przetwarzanie wstępne (podsumowanie sond i normalizację)
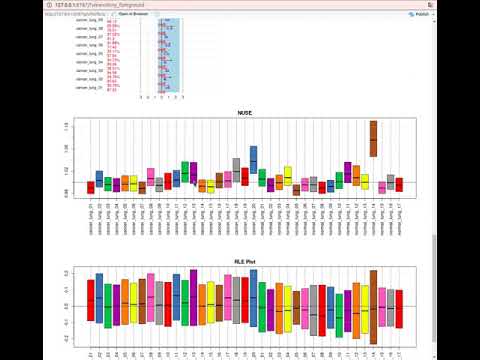
- Kontrolę jakości
- Wizualizację danych
- Obliczenie statystyk z ewentualną korektą wartości p
- Typowanie istotnych genów zgodnie z zadanymi kryteriami
- Przedstawienie ewentualnych wzorców
- Pobranie danych po normalizacji oraz wykonanych statystyk