FAQ
- General
- Errors
- Input
-
Output
- What should I do with incomplete genome?
- Why are there so many *path_sequence.fasta files?
- What are the differences between *1.path_sequence.fasta and *2.path_sequence.fasta?
- How does GetOrganelle choose the start point and strand for a circular genome?
- Why "circular genome" or "not circular genome" for embplant_nr/fungus_nr?
- What is the coverage of the assembly?
- How to generate the coverage plot across the assembled organelle genome?
Given that get_organelle_from_reads.py takes an input (or default) seed/label sequence, is it still a de novo assembler?
Yes, it is de novo.
The input seed sequence is only used as a starter for recruiting more overlapped reads. The recruited reads will further enrich themselves with more overlapped reads without the starter in the following extending rounds. Then all recruited reads will be de novo assembled into an assembly graph.
The label database is only used to improve distinguishing among plastome/mitogenome/nuclear sequence in the assembly graph filtering and disentangling process.
It is totally different from the typical reference genome-guided approaches where aligned reads are stitched into the new genome sequence and where the new genome is reconstructed based on the reference sequence. Here the organelle genome is totally de novo assembled from the recruited actual reads.
Currently, GetOrganelle does NOT offer a reference-guided assembly (see the answer above).
Instead, you may input a closely related organelle genome as the seed database (-s) and label database (--genes).
Using a custom seed database will be helpful in many situations (e.g., too few initial seed reads, imperfect parameter estimation, high sequencing heterogeneity); however, using a custom label database is NOT necessary/helpful unless 1) the target organelle is too diverged from the default label database (e.g., animal/fungal mitogenome), or other special purposes.
Although using custom databases is NOT typical for chloroplast genome assembly, let us use the chloroplast genome assembly as the example for reverse psychology fun:
-
If you want to use NC_047400 and NC_047059 as the custom seed database, what you need to do is simply download the fasta files from Genbank, cat them into one fasta file (e.g., legume_cp.fasta), and run GetOrganelle:
cat NC_047400.fasta NC_047059.fasta > legume_cp.fasta get_organelle_from_reads.py -s legume_cp.fasta -1 forward.fq -2 reverse.fq -o plastome_output -R 15 -k 21,45,65,85,105 -F embplant_pt -
If you want to use a combination of NC_047400, NC_047059, and NC_047060 as both the custom seed database and the custom label database :
-
Download the gb format files of NC_047400, NC_047059, and NC_047060.
-
Making the seed database. Convert the gb file into a fasta file. You definitely have tons of ways to do this; here is just one option
# pip install biopython # you may need to install biopython for transeq.py wget https://raw.githubusercontent.com/Kinggerm/PersonalUtilities/master/transeq.py python transeq.py genbank fasta NC_047400.gb NC_047059.gb NC_047060.gb cat NC_047400.fasta NC_047059.fasta NC_047060.fasta > legume_cp.seed.fasta -
Making the label database. Extract the cds from the gb file with sequence headers in the form of
>name type - any_other_info_without_space, e.g.>rbcL CDS - NC_047400--Chamaecrista_mimosoides. You may do it by yourself or use the GetOrganelle script get_annotated_regions_from_gb.py, a script designed for extracting genes from Genbank-format organelle genome data (previously available at https://github.com/Kinggerm/PersonalUtilities and mentioned in Zhang et al. 2020).# pip install biopython # biopython is additionally needed for this script get_annotated_regions_from_gb.py NC_047400.gb NC_047400.gb NC_047400.gb -o legume.cds -t CDS --mix mv legume.cds/gene/gene.fasta legume_cp.label.fasta -
run GetOrganelle using the seed database (NC_047400.seed.fasta) and label database (NC_047400.label.fasta)
get_organelle_from_reads.py -s legume_cp.seed.fasta --genes legume_cp.label.fasta -1 forward.fq -2 reverse.fq -o plastome_output -R 15 -k 21,45,65,85,105 -F embplant_pt
-
The idea of mapping and extending could be applied to recruiting loci/subgenomes other than listed organelle types (embplant_pt, embplant_mt, etc.).
Besides the usual GetOrganelle options, the relevant options you need for assembling custom loci are listed as follows:
-
-s seed.fasta(required) Used for initial recruitment, the fileseed.fastacould be one single sequence or a ton of sequences, preferably with sequences from closely related taxa included. Ideally, the result will be the set of the loci inseed.fasta. For example, if you have five completerbcLsequences and two completetrnL_UAAsequences inseed.fasta, the resulting assembly should be onerbcLand onetrnL_UAA. There is no sequence header preference forseed.fasta. Masking the repeats withNcould alleviate the computational burden from irrelevant reads. -
--genes label.fasta(required) Used for labeling and filtering contigs from the assembly, the filelabel.fastacould be similar or even the same toseed.fasta, except that the sequence headers are preferably like>name type - any_other_info_without_space, e.g.>rbcL gene - Amborella_AJ506156_2. -
-F anonym(recommended). # There may be some bugs in some versions, e.g., 1.7.3.3, which are now fixed in 1.7.3.5. -
--max-extending-len 100(conditional) Use this option to limit the read extending length approximately. The value choice (e.g., 100) largely depends on the gap/diverged bases you want GetOrganelle to fill in. -
-P 0(conditional) This should be chosen on ANY of the following cases, 1) your target has only one or a few copies compared to the nuclear genome, 2) all of your target loci have relatively short lengths (e.g., < 3000 bp).
Relevant posts: Discussion309
Recently, after installing GetOrganelle using conda, some users may be encountered the error bowtie2 cannot be found. Meanwhile, when they tested bowtie2 in the terminal, they got similar errors like:
/path-to/bin/bowtie2-align-s: error while loading shared libraries: libtbb.so.2: cannot open shared object file: No such file or directory. Also see issue 85.
This was caused by the most recent version of TBB (v2021.1.1), for which you may solve this by:
conda install tbb=2020.2
2021-04-13 11:47:46,760 - ERROR:
Time loading reference: 00:00:00
Time loading forward index: 00:00:00
Time loading mirror index: 00:00:00
Error: Read A12345:678:BCDE9FGHI:0:1234:5678:90123 1:N:0:TTAATTAA+CCGGCGGC has more quality values than read characters.
terminate called after throwing an instance of 'int'
(ERR): bowtie2-align died with signal 6 (ABRT) # or (ERR): bowtie2-align exited with value ##
This Read .. has more quality values than read characters error of Bowtie2 was mainly caused by the corruption of the input reads file.
Please use md5sum or shasum to ensure data integrity during every data transfer process.
If the original intact reads file(s) is missing, you may try to remove the garbled lines from the current corrupted file
and make it in the correct four-lines-per-sequence FASTQ format.
You can locate the garbled lines using grep, e.g gunzip -c input.fq.gz | grep -n "A12345:678:BCDE9FGHI:0:1234:5678:90123"
This is usually caused by insufficient system memory during reads recruitment or SPAdes assembly.
If this is caused before calling SPAdes, you may consider
- use a larger
-wif the estimated word size looks too small. - use the
--memory-saveflag, which is equivalent to--out-per-round -P 0 --remove-duplicates 0, to reduce memory usage. - use fewer reads with caution (can potentially reduce the success rate of completeness and accuracy)
- increase the available memory of the running environment
else if this is caused during SPAdes assembly, you have to consider
- use fewer reads with caution (can potentially reduce the success rate of completeness and accuracy)
- increase the available memory of the running environment
SPAdes is known to be not compliant with conda partially because the developers do not want to, and it is environmentally sensitive. I don't know if there is a list of which versions of SPAdes and what environments are compatible.
The current solution for most SPAdes-issue users is installing SPAdes from the source, according to the SPAdes developer. Remember to run spades.py --test to confirm. Users who installed GetOrganelle using conda should additionally uninstall the conda-installed spades or use --which-spades to guide GetOrganelle to use the correct version. Otherwise, the error will seem to stay.
Similar symptoms:
- WARNING: Pre-assembling failed
- WARNING: Assembling exited halfway
- ERROR: Assembling failed
- ERROR: Error with running SPAdes
- ERROR: Assembling failed
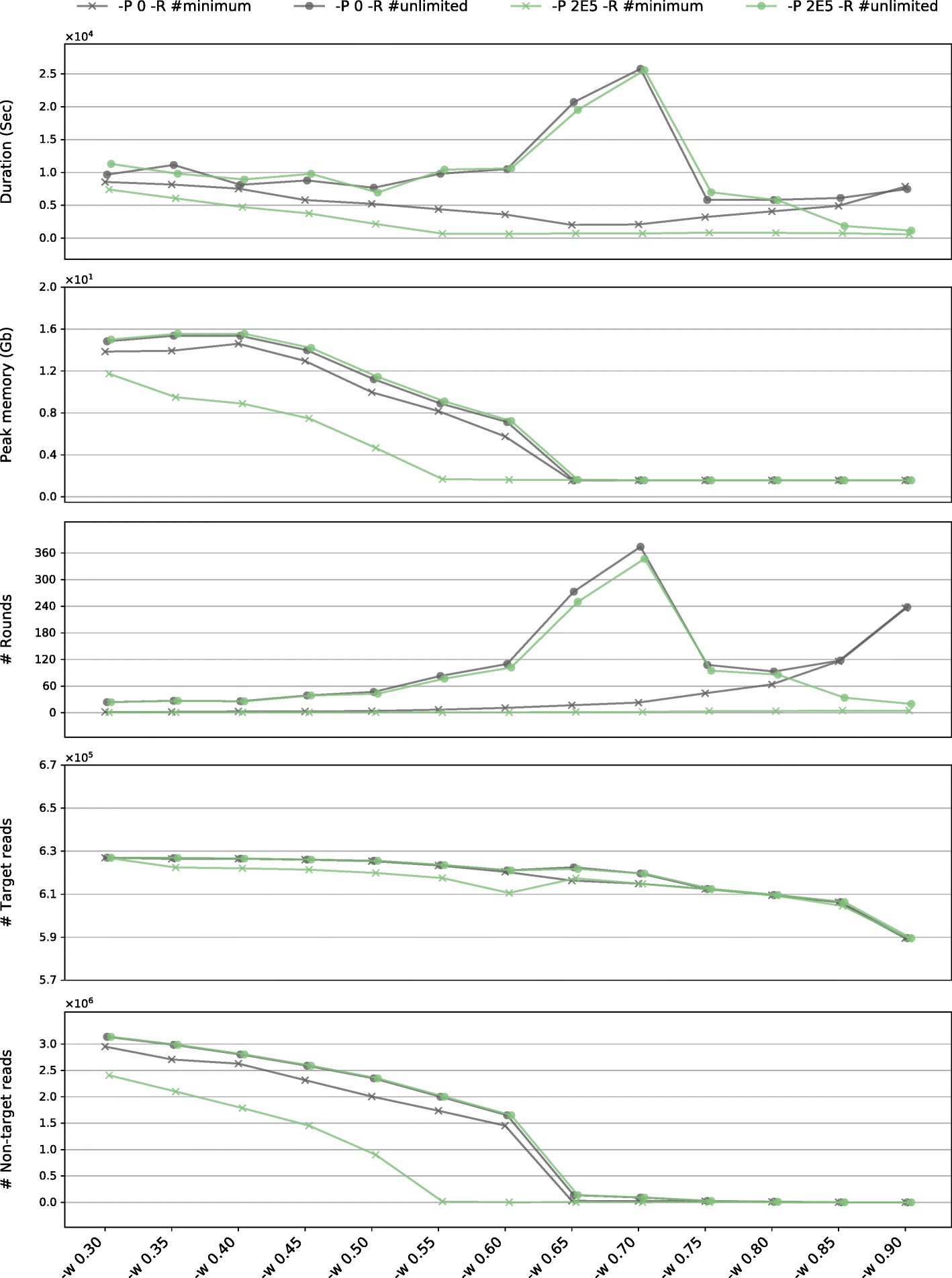
This answer is complementary to the Word size in GetOrganelle section in our GetOrganelle paper. Please refer to that section before reading below.
The value word size, like the kmer in assembly, is crucial to the feasibility and efficiency of this process. The best word size changes upon data and will be affected by read length, read quality, base coverage, organ DNA percent and other factors. By default, GetOrganelle would automatically estimate a proper word size based on the data characters. Although the automatically-estimated word size value does not ensure the best performance nor the best result, you do not need to adjust this value (-w) if a complete/circular organelle genome assembly is produced, because the circular result generated by GetOrganelle is highly consistent under different options and seeds. However, the word size values will significantly influence the computational burden in both memory and time (see GetOrganelle paper Fig 6 attached below). The automatically estimated word size may be screwy in some animal mitogenome data due to inaccurate coverage estimation, and in some plant mitogenome data due to an intrinsic large dataset requirement, for which you fine-tune it instead.
A good word size value should not only be helpful in yielding a complete organelle genome but also accommodate the computational resources we can afford. In general, we need to have an expectation of how many target-associated reads should be recruited. Because we are making the recruited reads include all target reads and inevitably include some non-target reads, the expected number of recruited reads is usually 1.5x to 4.x of organelle DNA percent, depending on the depths of nuclear, pt and mt, and on how much nuclear, pt, mt share the similarity. For example, if you have
2022-02-22 12:34:56,789, - INFO: Round 10: 80000000/80000000 AI 12345678 AW 123456789 Mem 43.21
as the last round of your latest trial of the read extension, you have the recruited ratio of 12345678/80000000=15.4% which is a reasonable number for plastome assembly, since the plastid DNA percent is around 5% (and can be variable). The word size of this run can be considered good, as long as the result is complete. And if the recruited ratio of a run is >30% for plastome in a real WGS dataset, it is likely wasting the computational resources and you should consider increasing the word size value. However, it is hard to tell the computational cost for a word size and a dataset prior to running.
Since get_organelle_from_reads.py is conducting de novo assembly, the input seed/label database have little impact on the final assembly as long as 1) the assembled organelle genome is complete, 2) the assembled organelle genome is the same type (e.g., pt vs. mt) to the input database, 3) the reads is not contaminated.
However, the seed/label database will strongly influence the completeness chance when 1) the coverage is very low or low in some regions, or 2) the target organelle genome has a very high substitution rate (e.g., animal/fungal mitogenomes). In these cases, using custom databases will increase the assembly completeness chance.
In short, input as many reads as you can, and get_organelle_from_reads.py will automatically estimate the number of reads/read pairs to use, which could be controlled by flags --max-reads and --reduce-reads-for-coverage.
There's no exact answer for all when it comes to how many are required. Different samples have different target read percentages. However, the default values for --max-reads are generally sufficient for most samples.
Some users complain about the running speed of GetOrganelle compared to NOVOPlasty. However, the default parameters prefer accuracy/success more than computational efficiency. Therefore, one may fine-tune the parameters below to significantly speed up the first run while still maintaining a better accuracy and success rate than NOVOPlasty:
-
-R, a smaller number of rounds (default: conditioning on-F) will reduce the extended (recruited) reads, therefore less (SPAdes) assembly time. -
--larger-auto-ws, use this flag to generate a slightly larger word size and less extended reads, therefore less assembly time. -
-t, a larger number of threads (default: 1) will speed up the dependencies (bowtie2, SPAdes, etc.) -
-J, a larger jump size (default: 3) will speed up the extending and reduce the extended reads -
-M, a larger mesh size (default: 2) will speed up the extending and reduce the extended reads -
--max-n-words, a smaller maximum number of words (default: conditional on-F) will terminate the extending earlier in some cases, therefore less extended reads and assembly time. -
--ignore-k, a larger threshold will skip more trivial slimming/disentangling runs. -
--fastis a short cut of using-R 10 -t 4 -J 5 -M 7 --max-words 3E7 --larger-auto-ws --disentangle-time-limit 360. As the default, this mode is an arbitrary choice with much less running time and a lower success rate (still high among available tools, though).
or fine-tune parameters combined with bove tunings according to a previous run on the same or a similar dataset:
-
-w, a larger word size (than the auto-estimated one) will generally speed up the entire run by reducing the extended reads given a fixed number of rounds.
Given that there could be a trade-off between success and speed, the above tunings may or may not reduce the success rate. However, as long as the resulting assembly is successful (complete circular), GetOrganelle generally generates consistent assemblies (Jin et al. 2020), which is great for those who want a quick sample scan.
Besides, parameters for animal mitogenome assembly have not been fine-tuned well yet to maximize neither the successful rate nor the computational efficiency (e.g., the auto-estimated word size may be too small). We would appreciate your feedback on your empirical datasets.
Before checking the instructions below, please use the recommended commands first and troubleshoot any ERROR (installation or running errors). For embplant_nr/fungus_nr assembly, please first refer to here before trying to improve.
-
If the assembly graph (*.fastg) is an organelle-sufficient graph.
Use Bandage to visualize the assembly graph (load both the *.fastg and *.csv) to confirm this. The organelle-sufficient assembly graph contains the target genome sequence, but GetOrganelle did not navigate the right path(s) for you. Generally, you do not need to adjust the parameters to rerun the same dataset, but modify the assembly graph or look for other evidence for disentangling.
1.1 If the
WARNINGisDisentangling timeoutand the organelle contigs in the graph are not inverted-repeat-rich, please increase the time limit, e.g.--disentangle-time-limit 7200.1.2 If the graph is inverted-repeat-rich, please try to resolve the graph using additional evidence, e.g., mapping with long-insert-sized paired reads or long reads that could spin the repeat region.
1.3 If the graph has any contig that has exceptionally high coverage (e.g., ten times the average) and GetOrganelle failed to estimate the multiplicity, you can try to increase the maximum kmer value (127 for SPAdes) or do the semi-manual disentanglement in Bandage (see here or here).
1.4 If the graph contains many parallel contigs, which may be caused by a mixture of multiple resources (contamination, horizontal transfer between different organelle genomes, or heteroplasmy). It would help if you did some semi-manual completion, e.g., manually remove some non-target contigs and then do the completion using
get_organelle_from_assembly.py.- contamination. Tips: 1) two sets of complete plastome could be found; 2) graph networks around the IR boundaries.
- horizontal transfer between different organelle genomes. Tips: 1) Compared to the true target contig, the transferred contig usually has 1) different blast hit in the Genbank nucl database; 2) the genes in the transferred contig are more likely to be pseudogenized; 3) the transferred sequence usually have rearrangements which leads to abnormal bridging between the true target contigs.
- heteroplasmy.
-
If the assembly graph (*.fastg) is NOT organelle-sufficient.
2.1 If the graph is near complete (e.g., 1~4 contigs/scaffolds), try rerun
get_organelle_from_reads.pywith the following parameter adjustments (helpfulness-decreasingly ranked; most helpful options are marked with ^):- ^ reducing word size (
-w) if your working environment could supply more memory. If no word size was given for the first run, it would be automatically estimated by the program and logged in theget_org.log.txt, where you can find the value to start reducing. For example, if the estimated word size was 105, I would try a 95 next. A reasonable value is usually between 65 and 105. Reducing word size will be more helpful when AW/AI seems stable/barely changed as rounds increase in the old run. - ^ increasing input reads (
--reduce-reads-for-coverageor--max-reads) if reads were reduced by any of these two options. - ^ using a closely-related organelle genome as the seed (
-s) if the target genome is animal mitogenome (highly recommended), or the quality of the reads is poor, or target coverage is highly uneven. For animal genome assembly or cases without closely-related seed, using the output of the previous run as the seed for a second run will be a good choice. - increasing rounds (
-R). The suggested number of rounds is sufficient for most samples unless your sample has extremely shallow coverage and the seed is highly divergent from the target. - using wider and denser k-mer gradient (
-k) - narrowing extending steps (
-J/-M) - looking for help at Discussion with the get_org.log.txt and the assembly graph (*.fastg or *.png with length/depth/name turned on) attached.
Besides parameter fine-tuning, there are other two different approaches to try: you may try using the script make_batch_for_iteratively_mapping_assembling.py to generate a bash file to run iteratively Bowtie2-mapping and SPAdes-assembling; alternatively (if you have sufficient computational resources), you can assemble the raw reads directly using SPAdes, then use
get_organelle_from_assembly.pyto extract the target organelle genome sequence.2.2 If the graph is highly fragmented, you can still try the above parameter adjustment (see 2.1), but the completeness chance will be meager. To achieve a circular sequence with gaps/overlaps (* does not bring any new bases but just for more accessible downstream analysis), you can use
join_spades_fastg_by_blast.pyto modify theassembly_graph.fastg.extend-*.fastgfile (Graph A), insert gaps in-between contigs, and created a gap-containing organelle-sufficient graph (Graph A') with a reference genome, then useget_organelle_from_assembly.pyto extract the gap-containing "complete" organelle genome from theGraph A'. Manual graph checking/pruning using Bandage between above steps may help work out. You can also do the above steps for multiple iterations. - ^ reducing word size (
If you are assembling plastome with IRs and get two *fasta files, please see the answer to the next question.
Otherwise, those *path_sequence.fasta files are caused by other repeats, accounting for the assembling uncertainties. As a de novo organelle genome assembler, GetOrganelle exports all possible configurations by default.
The Fig 2 of GetOrganelle paper (attached below) expressed this "all possible configurations" idea.
In this Fig 2 plastome case, the two symmetrical configurations (inside the green dashed-line box) out of all six configurations are more likely to be the two real plastome structures.
The two *.fasta files with those two symmetrical configurations will be marked with repeat_pattern1 in their file names, e.g. embplant_pt.K105.complete.graph1.1.repeat_pattern1.path_sequence.fasta and embplant_pt.K105.complete.graph1.2.repeat_pattern1.path_sequence.fasta.

Another solvable case comes from the SC and the IR region sharing short repeats; in that case, repeat_pattern may or may not be seen in the output fasta file names. The detailed solutions are described in issue-86.
In other cases (animal mitochondrial genome or other organelle genome types),
when there's no clue of what kind of configurations are more realistic,
you have to keep the uncertainties (e.g., using the contigs of the GFA file for downstream analysis),
or use other data sources, e.g., the library of read pairs or long reads spanning over the repeat regions for repeat resolution.
What are the differences of complete*.1.path_sequence.fasta and complete*.2.path_sequence.fasta output files?
If you assemble the plastome with IRs, the assembly result should be two equimolar isomeric sequences. Both of them are right and coexist in the plant (Palmer 1983; Walker et al. 2015). For publication, people tend to use just one of them. You can pick the configuration of which the SSC region is in the same direction as most of your data or outgroup, which makes downstream annotation/analysis easier.
Fig 1. Typical Assembly Graph of a Plastome (Issue 22)

Similar questions:
-
What shall I do if there's "WARNING: Please check the final result to confirm whether they are simply flip-flop configurations"(Issue 5).
-
Why the SSC/LSC region is different from assemblies from another source?
Because most organelle genomes are double-stranded and circular, there is no way to define an explicit start and end for the circular sequence in a short-read-based de novo assembly, meaning that one may randomly reseed the start point (so-called "cut-and-paste") even switch the strand without changing the biological meaning of that sequence. Nevertheless, people have a specific tradition of the start point and the strand for each organelle genome type. Please refer to a number of published annotated organelle genomes in your field.
Currently, the general principle that GetOrganelle uses to pick the strand and the start point (cutting point in a circular genome) is starting with the orf-richest strand of the longest single copy contig.
Besides, two extra modifications were made for embplant_pt: 1) the searching will expand to an entire continuous single copy region rather than just a single copy contig, namely the longest single copy region; 2) the strand will be the orf-least strand of that region.
If you have achieved a complete chloroplast genome with IRs but want to reverse the output LSC region, you may use
get_organelle_from_assembly.py --reverse-lsc --no-slim -F embplant_pt -g the_previous_output.gfa -o new_output_1
get_organelle_from_assembly.py --no-slim -F embplant_pt -g the_previous_output.gfa -o new_output_2
You may find your ideal result in one of the new_output_1 and new_output_2.
Similar questions:
- What shall I do if the LSC region is in a reverse direction from a closely-related published chloroplast genome?
- Why is the start point of my assembled genome different from the reference/published organelle genome?
The nuclear ribosomal DNAs generally formed tandem repeats in the following units: -(ETS-18S-ITS1-5.8S-ITS2-28S-IGS)-.
Ideally, if every two units are connected end-to-end, it can be represented by a circular sequence.
Practically, short reads will also generate a circularized assembly graph when all regions are concertedly evolved. Therefore you may get the "circular genome" in GetOrganelle 1.7*.
However, please keep in mind that in many cases, IGS will not be completely concerted, creating complicated bubbles and knots in the assembly graph. Therefore, in this commonly-seen case, GetOrganelle will generate multiple different circular sequences or will not generate circular results but linear scaffolds (ideally one scaffold with the length of 6~10 Kbp). Even if concerted ("circular genome" generated), the IGS region can barely be aligned to closely related species, therefore useless for many downstream analyses.
Besides, ITS will not be completely concertedly evolved in many cases, creating complicated bubbles in the assembly graph.
The coverage of the assembly (C) will be logged in the final block of the log file, such as:
20YY-MM-DD HH:MM:SS,SSS - INFO: Average embplant_pt base-coverage = 234.5
In short, 234.5x is what you need to report as the assembly coverage.
This value is actually the commonly-used base coverage, which is derived from the k-mer coverage contained in the final assembly graph using the formula C=Ck*L/(L−k+1), where Ck is the k-mer coverage, k is the kmer size, L is the read length (also see velvet manual p14).
Yet still, there are a few more things people may want to know.
-
The
total number of reads that GetOrganelle used to achieve the organelle genome(N) was a little tricky. Please note thatNwill be <= thenumber of raw reads, and be >= theexact number of reads to form the organelle genome(O, see point 4 below). GetOrganelle will do no, one or two rounds of data reducing depending on the estimated target coverage (to force no reducing, see--max-readsand--reduce-reads-for-coverage). TheNafter the first reducing process (or no reducing) will be logged inside the block ofPre-reading fastqin the form of20YY-MM-DD HH:MM:SS,SSS - INFO: Pre-reading fastq ... ... 20YY-MM-DD HH:MM:SS,SSS - INFO: Reads used = A1+A2 20YY-MM-DD HH:MM:SS,SSS - INFO: Pre-reading fastq finished., where the sum of
Anumbers is theN, i.e., the number of reads for downstream analysis. If the estimated target coverage is still larger than a given threshold, GetOrganelle will do a second reducing process. The updatedNafter the second reducing process will be logged inside the block ofChecking seed reads and parametersin the form of20YY-MM-DD HH:MM:SS,SSS - INFO: Checking seed reads and parameters ... ... 20YY-MM-DD HH:MM:SS,SSS - INFO: Reads reduced to = B1+B2 ... 20YY-MM-DD HH:MM:SS,SSS - INFO: Checking seed reads and parameters finished., where the sum of
Bnumbers is theN, i.e., the number of reads for downstream analysis.Thus, the
Nis-
sum(
B), ifReads reduced to = B1+B2is observed in the block ofChecking seed reads and parameters -
sum(
A), otherwise
-
-
The
extended*.fqfile(s) in the output folder is just the intermediate file(s) that GetOrganelle passed to SPAdes, which still contains non-target reads. The number(s) of reads inextended*.fqfile(s) should never be considered in answeringhow many reads were usedquestions. -
The
percentage of the target organelle genome in the WGS data(P) can be calculated asP=S*C/(N*L), whereSis the assembled organelle genome size,Cis the coverage of the assembly as calculated above,Nis the number of reads that GetOrganelle used as calculated above,Lis the read length. -
There is no direct information for the
exact number of reads to form the organelle genome(O) because the target organelle genome is a subset of the WGS sequencing and the assembly results from an integrated extending+assembly+graph-filtering process. However,Ocan be approximately calculated as the portion of the target organelle genome in the used reads, i.e.,O≈P*N=S*C/L.
Similar questions:
- How many reads were used to conduct the organelle genome assembly
- What is the percentage of the organelle genome in the WGS data
To generate the coverage plot, you may use
evaluate_assembly_using_mapping.py -f assembly.fasta -1 extended_1.fq -2 extended_2.fq -o output_dir -c yes --draw
For example, given reads file SRR5602592 and its assembled chloroplast genome embplant_pt.K127.complete.graph1.1.path_sequence.fasta, using the following command can yield the example plot. In this example, the original reads were used for mapping. However, using the extended_*.fq data would produce a similar result with a faster processing time.
evaluate_assembly_using_mapping.py -1 SRR5602592_1.fastq.gz -2 SRR5602592_2.fastq.gz -o auto--SRR5602592-plastome--evaluated -f embplant_pt.K127.complete.graph1.1.path_sequence.fasta -t 1 --continue --draw --plot-title SRR5602592-GetOrganelle --plot-subtitle "Arguments: -R 10 -t 1 -k 21,55,91,127 Result status: circular genome"

Find more examples in the benchmark tests. Also, see more options using
evaluate_assembly_using_mapping.py -h