![]()
STREAMLINE is an end-to-end automated machine learning (AutoML) pipeline that empowers anyone to easily train, interpret, and apply a variety of predictive models as part of a rigorous and optionally customizable data mining analysis. It is programmed in Python 3 using many common libraries including Pandas and scikit-learn.
The schematic below summarizes the automated STREAMLINE analysis pipeline with individual elements organized into 9 phases.
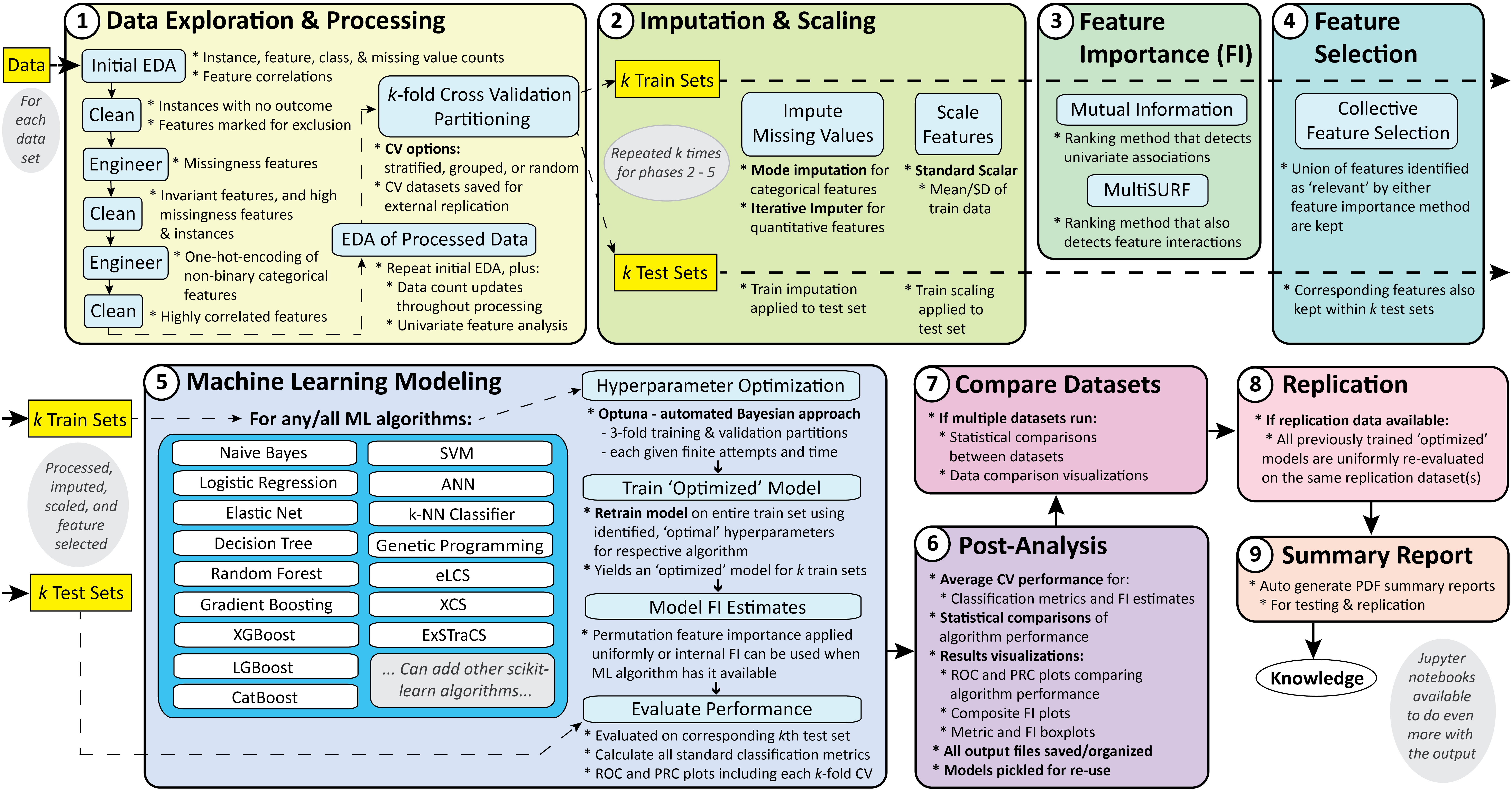
-
Detailed documentation of STREAMLINE is available here.
-
A simple demonstration of STREAMLINE on example biomedical data in our ready-to-run Google Colab Notebook here.
-
A video tutorial playlist covering all aspects of STREAMLINE is available here

The goal of STREAMLINE is to provide an easy and transparent framework to reliably learn predictive associations from tabular data with a particular focus on the needs of biomedical data applications. The design of this pipeline is meant to not only pick a best performing algorithm/model for a given dataset, but to leverage the different algorithm perspectives (i.e. biases, strengths, and weaknesses) to gain a broader understanding of the associations in that data.
The overall development of this pipeline focused on:
- Automation and ease of use
- Optimizing modeling performance
- Capturing complex associations in data (e.g. feature interactions)
- Enhancing interpretability of output throughout the analysis
- Avoiding and detecting common sources of bias
- Reproducibility (see STREAMLINE parameter settings)
- Run mode flexibility (accomodates users with different levels of expertise)
- More advanced users can easily add their own scikit-learn compatible modeling algorithms to STREAMLINE
See the About (FAQs) to gain a deeper understanding of STREAMLINE with respect to it's overall design, what it includes, what it can be used for, and implementation highlights that differentiate it from other AutoML tools.
-
At present, STREAMLINE is limited to supervised learning on tabular, binary classification data. We are currently expanding STREAMLINE to multi-class and regression outcome data.
-
STREAMLINE also does not automate feature extraction from unstructured data (e.g. text, images, video, time-series data), or handle more advanced aspects of data cleaning or feature engineering that would likely require domain expertise for a given dataset.
-
As STREAMLINE is currently in its 'beta' release, we recommend users first check that they have downloaded the most recent release of STREAMLINE before use. We are actively updating this software as feedback is received.
The most recent publication on STREAMLINE (release Beta 0.3.4) with benchmarking on simulated data and application to investigate obstructive sleep apena risk prediction as a clinical outcome is available as a preprint on arxiv here.
The first publication detailing the initial implementation of STREAMLINE (release Beta 0.2.4) and applying it to simulated benchmark data can be found here, or as a preprint on arxiv, here.
See citations for more information on citing STREAMLINE, as well as publications applying STREAMLINE and publications on algorithms developed in our research group and incorporated into STREAMLINE.
STREAMLINE can be run using a variety of modes balancing ease of use and efficiency.
- Google Colab Notebook: runs serially on Google Cloud (best for beginners)
- Jupyter Notebook: runs serially/locally
- Command Line: runs serially or locally
- Locally, serially
- Locally, cpu core in parallel
- CPU Computing Cluster (HPC), in parallel (best for efficiency)
- All phases can be run from a single command (with a job monitor/submitter running on the head node until completion)
- Each phase can be run separately in sequence
See the documentation for requirements, installation, and use details for each.
Basic installation instructions for use on Google Colab, and local runs are given below.
There is no local installation or additional steps required to run STREAMLINE on Google Colab.
Just have a Google Account and open this Colab link to run the demo (takes ~ 6-7 min): https://colab.research.google.com/drive/14AEfQ5hUPihm9JB2g730Fu3LiQ15Hhj2?usp=sharing
Install STREAMLINE for local use with the following command line commands:
git clone --single-branch https://github.com/UrbsLab/STREAMLINE
cd STREAMLINE
pip install -r requirements.txt
Now your STREAMLINE package is ready to use from the STREAMLINE folder either
from the included Jupyter Notebook file or the command line.
Included with this pipeline is a folder named DemoData including two small datasets used as a demonstration of
pipeline efficacy. New users can easily test/run STREAMLINE in all run modes set up to run automatically on these datasets.
A complete list of STREAMLINE Parameters can be found here.
We make no claim that this is the best or only viable way to assemble an ML analysis pipeline for a given classification problem, nor that the included ML modeling algorithms will yield the best performance possible. We intend many expansions/improvements to this pipeline in the future. We welcome feedback, suggestions, and contributions for improvement.
We welcome ideas, suggestions on improving the pipeline, code-contributions, and collaborations!
-
For general questions, or to discuss potential collaborations (applying, or extending STREAMLINE); contact Ryan Urbanowicz at ryan.urbanowicz@cshs.org.
-
For questions on the code-base, installing/running STREAMLINE, report bugs, or discuss other troubleshooting issues; contact Harsh Bandhey at harsh.bandhey@cshs.org.






The development of STREAMLINE benefited from feedback across multiple biomedical research collaborators at the University of Pennsylvania, Fox Chase Cancer Center, Cedars Sinai Medical Center, and the University of Kansas Medical Center.
The bulk of the coding was completed by Ryan Urbanowicz, Robert Zhang, and Harsh Bandhey. Special thanks to Yuhan Cui, Pranshu Suri, Patryk Orzechowski, Trang Le, Sy Hwang, Richard Zhang, Wilson Zhang, and Pedro Ribeiro for their code contributions and feedback.
We also thank the following collaborators for their feedback on application of the pipeline during development: Shannon Lynch, Rachael Stolzenberg-Solomon, Ulysses Magalang, Allan Pack, Brendan Keenan, Danielle Mowery, Jason Moore, and Diego Mazzotti.
Funding supporting this work comes from NIH grants: R01 AI173095, U01 AG066833, and P01 HL160471.